Article Text

Abstract
Toxic neuropathies result from exogenous substances damaging the peripheral nerves. There are numerous causes, including prescribed and recreational drugs, heavy metals, industrial agents and biological toxins. Timely recognition of these neuropathies gives better outcomes, as they usually improve or stabilise once the toxin is removed. Most toxic neuropathies are axonal, length-dependent and sensory predominant, although some have significant motor involvement or can present acutely or subacutely. Here, we outline our clinical approach and discuss the major causes of toxic neuropathy, while emphasising the clinical and neurophysiological features and the neuropathy phenotype. We also include an update on newer medications that can cause neuropathy, including immune checkpoint inhibitors and BRAF/MEK inhibitors.
- NEUROPATHY
- NEUROONCOLOGY
- NEUROPHYSIOLOGY
- NEUROTOXICOLOGY
- CLINICAL NEUROLOGY
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
Statistics from Altmetric.com
Introduction
Toxic neuropathies occur when chemicals, pharmaceuticals or dietary substances cause damage or dysfunction to peripheral nerves. While many of the individual causes are rare, toxic neuropathies collectively are not uncommon, with chemotherapies and alcohol as the most frequent causes. The many other causes include recreational drugs, heavy metals and dietary and industrial toxins. Removing the offending substance can lead to clinical improvement and, given the available specific treatment for some toxins, early and correct identification of the relevant toxin is important. Neurologists should consider screening for exposure to appropriate neurotoxins according to the neuropathy phenotype. Being aware of the particular neuropathy pattern associated with each toxin helps with clinical recognition and subsequent appropriate investigation and management. This knowledge can also help other specialists, such as oncologists and haematologists, to distinguish toxic neuropathies (eg, because of chemotherapy) from other disease-related causes of a neuropathy, such as light chain (AL-) amyloidosis.
While most toxic neuropathies present as a typical ‘length-dependent’ predominantly sensory axonopathy, some toxins can cause other patterns such as a motor predominant neuropathy or a polyradiculoneuropathy. In these more unusual cases, it is particularly important to exclude other causes, as the toxin exposure can be coincidental. Some toxins also affect other parts of the nervous system including muscle, optic nerve and central nervous system (CNS), or can affect other organs, all or any of which can provide diagnostic clues (table 1).
Clinical features of toxic neuropathies
General approach
History and examination
The history should focus on the motor (eg, weakness, cramps), sensory (numbness, pain) and autonomic (such as presyncope, erectile difficulty or urinary urgency) symptoms. Systemic symptoms such as nausea, weight loss or skin changes are common with many toxins, occasionally being pathognomonic and diagnostic, and these should also be sought. A drug history should include current and previously prescribed and over-the-counter medications, including nutritional supplements, as well as recreational drugs. A thorough social history is important and should include occupation, travel, diet and possible environmental exposures. The time course is particularly informative; the symptoms should start at a timepoint consistent with the known onset of neuropathy with the purported toxin and improvement or stabilisation should occur when the drug or toxin is removed (with a few exceptions). It is thus often possible to confirm a toxic neuropathy only retrospectively.
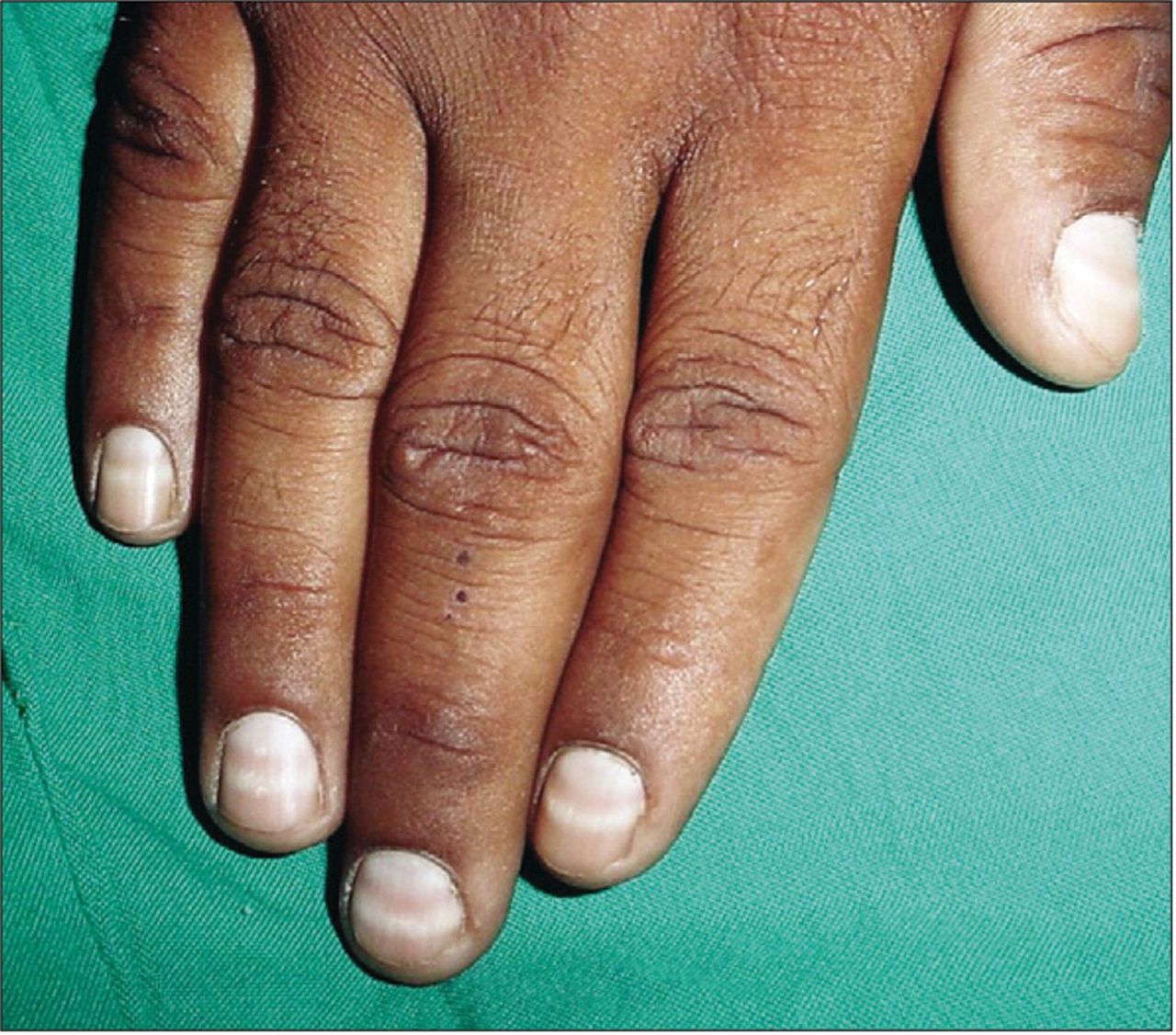
In addition to the standard neurological examination for a peripheral neuropathy and its linked neuropathic features, a careful systemic examination including the skin, nails and hair can identify ‘tell-tale signs’ of toxicity: for example, hyperkeratosis and Mees’ lines (on fingernails, figure 1) with arsenic and thallium toxicity.

Mees’ lines. From: Chauhan et al. 58 Reproduced with permission from Elsevier.
Investigations
Nerve conduction studies complement the clinical assessment to help to classify the pattern of neuropathy—axonal vs demyelinating (more correctly, conduction slowing), sensory vs sensorimotor vs motor—and the neurophysiological pattern may give a clue to diagnosis. In addition to a standard ‘neuropathy screen’ in blood workup, clinicians should consider investigating for particular toxic neuropathies in certain situations (table 1).
Chemotherapy-induced peripheral neuropathy
The most common phenotype in chemotherapy-induced peripheral neuropathy is a length-dependent sensory predominant axonal neuropathy, but there are some exceptions. This complication is very common with neurotoxic chemotherapy regimens, occurring in approximately 30%–40% of those exposed.1 Risk factors for neurotoxicity include increasing age, diabetes, obesity, high alcohol intake, pre-existing neuropathy, anaemia and low pretreatment concentrations of vitamin D, magnesium and albumin.2 Neurotoxicity is a major dose-limiting side effect in cancer management. Reducing the chemotherapy dose can reduce survival from the malignancy,3 and a neuropathy can adversely impact quality of life in survivors. Chemotherapy-induced peripheral neuropathy usually begins weeks to months after starting treatment and then stabilises or improves once treatment ceases, although the phenomenon of coasting (continued worsening after stopping chemotherapy for 2–3 months) is well documented, particularly with platinum compounds. The most helpful clinical feature in differentiating a toxic neuropathy from another cause of peripheral nerve symptoms in the cancer cohort is the temporal relationship to drug exposure. Metabolic, nutritional and disease-related (vasculitis, paraneoplastic neuropathies, AL amyloidosis) neuropathies must also be considered in cancer patients. Table 2 summarises the chemotherapy drugs causing neuropathy and their clinical features.
Chemotherapy drugs commonly associated with peripheral neuropathy
Platinum chemotherapies
The platinum agents cisplatin, carboplatin and oxaliplatin can form platinum adducts with nuclear and mitochondrial DNA in the dorsal root ganglia, leading to apoptosis.1 Cisplatin and oxaliplatin are neurotoxic, and while carboplatin may cause neuropathy, this is less common. As the pathology is primarily in the dorsal root ganglia, platinums usually cause a sensory neuronopathy and present with sensory symptoms or sensory ataxia. Cisplatin neuropathy not uncommonly worsens for a couple of months after stopping the treatment, a phenomenon known as coasting. Cisplatin can also cause ototoxicity, with tinnitus, hearing loss and (less commonly) vestibular failure.4 Oxaliplatin causes an acute neural hyperexcitability syndrome in over 90% of patients,5 usually resolving within several days after each infusion according to the summative literature. In our experience, however, subjective complaints sometimes last for a week or longer. During this time, patients can experience paraesthesiae (particularly of the hands and face), cold intolerance, cramps, fasciculation and throat tightness, among other symptoms. The acute hypersensitivity syndrome is likely to be a channelopathy and while it does not represent nerve damage per se, longer and more severe acute symptoms have been associated with an increased risk of chronic neuropathy, and it remains possible that the hyperexcitability syndrome could lead to chronic neuropathy.6 Chronic oxaliplatin neuropathy is similar to that caused by cisplatin, and while coasting can occur, it is less common.
Taxanes
Taxanes disrupt normal microtubule function, leading to impaired axonal transport, which may underlie their neurotoxic effects.1 Taxane-induced neuropathy is primarily sensory, with less common and less severe motor involvement,5 and patients classically present with length-dependent sensory loss and neuropathic pain. There is also an acute pain syndrome comprising arthralgia and myalgia (and sometimes myopathy) seen in around 10%–30% of patients, starting 1–2 days after an infusion and lasting around 3–5 days.7 Paclitaxel comes with the highest risk of chemotherapy-induced peripheral neuropathy, followed by docetaxel, while cabazitaxel is significantly less neurotoxic.
Vinca alkaloids
The vinca alkaloids vincristine, vinblastine and vinorelbine are primarily used in haematological cancers, with vincristine the most neurotoxic of the group. These agents impair microtubule function and can cause a sensorimotor neuropathy, often with distal weakness.1 Symptoms frequently begin early in the chemotherapy course, and coasting can also occur.5 Constipation is common, and patients can develop generalised dysautonomia and occasionally cranial nerve involvement.5 Vinca alkaloids are contraindicated in patients with Charcot-Marie-Tooth disease type 1A as they can cause an acute and severe neuropathy.8
Proteasome inhibitors
Bortezomib causes neuropathy by mechanisms that are not fully worked out or understood, but may involve neuroinflammatory processes.9 Bortezomib-induced neuropathy is most commonly sensory and usually painful, which may be a result of a predilection for small nerve fibre (pain and temperature) involvement.5 Patients experience sharp or burning pain in the extremities, which can be debilitating, often necessitating reducing the dose or stopping the drug. Autonomic involvement has been reported,5 as have other neuropathy phenotypes including distal motor weakness and multifocal motor neuropathy-like presentations.10 Other proteasome inhibitors (carfilzomib, ixazomib) are much less likely to cause neuropathy, and bortezomib given subcutaneously is less likely to cause neuropathy than when given intravenously.1
Immunomodulatory drugs
Thalidomide was once commonly used in multiple myeloma treatment regimens, though in recent years has mainly been superseded by newer agents due to the common occurrence of sensory neuropathy and other side effects. Thalidomide is antiangiogenic and may cause neuropathy by reducing blood supply to peripheral nerves.5 This usually occurs with cumulative doses above 20 g and may occur in more than 80% of recipients at this dose.5 The newer agents lenalidomide and pomalidomide are more potent and better tolerated, with much lower rates of neurotoxicity. Lenalidomide is a first-line treatment for polyneuropathy, organomegaly, endocrinopathy, M-protein and skin changes (POEMS) syndrome and is very unlikely to worsen the neuropathy of this condition.
Brentuximab vedotin and antibody–drug conjugates
Antibody–drug conjugates are a newer class of antineoplastic drugs consisting of an anti-tumour-associated antigen monoclonal antibody linked to a cytotoxic molecule. The drug enters the cell by binding to the antigen, and the cytotoxic agent is released within the cell. While this class of medications is carefully targeted to tumour cells, occasionally there can be off-target effects, including neuropathy. Brentuximab vedotin contains an anti-CD30 monoclonal antibody conjugated to a potent antimicrotubule agent and is used in Hodgkin’s lymphoma and other haematological malignancies. Over half of recipients develop a length-dependent, large fibre predominant sensory axonal neuropathy, causing distal sensory loss and imbalance.11 Most of these improve after stopping the drug. There are also other antibody–drug conjugates with described neurotoxic effects in use, with many more in clinical trial settings.
Immune checkpoint inhibitors
In recent years, immune checkpoint inhibitors have become an important part of therapy for increasing numbers of cancers. Immune checkpoints are nodal ‘stop points’ in immune pathways that prevent excessive immune activation. Inhibition of immune checkpoints leads to increased T-cell activation, with the purpose of an increased antitumour response. However, unrestricted T-cell activation comes with a risk of autoimmune side effects. Immune checkpoints targeted for inhibition include cytotoxic T lymphocyte activation (CTLA)-4 (eg, ipilimumab) and programmed death (PD)-1 (eg, nivolumab, pembrolizumab). Neurological autoimmune side effects are relatively uncommon and tend to affect the peripheral more than the central nervous system.12 Rates of neurological toxicity are 1%–4% of patients treated with CTLA-4 inhibitors, 3%–6% treated with PD-1 inhibitors and 12%–14% treated with drugs in combination.13 14 There are many different neuropathy phenotypes reported with immune checkpoint inhibitors, with acute/subacute polyradiculoneuropathy the most common.12 Despite some clinical similarities to Guillain-Barré syndrome or chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) with regard to temporal onset and anatomical distribution of deficits, immune checkpoint inhibitor-related immune neuropathies have a different pathology and should be treated with high-dose corticosteroids rather than intravenous immunoglobulin. Plasma exchange may be useful in severe cases. The neuropathy can recur if the immune checkpoint inhibitors are restarted, or if immune suppression is reduced too quickly.12
BRAF and MEK inhibitors (eg, dabrafenib, trametinib)
Mutations in the BRAF gene are a key player in several tumours, including melanoma, non-small cell lung cancer and anaplastic thyroid cancer. Mitogen-activated protein kinase kinase (MAP2K or MEK) is a downstream signalling target of BRAF, and inhibitors of these two key signalling proteins in the mitogen-activated protein kinase pathway are currently in clinical use. Peripheral neuropathy is emerging as an uncommon complication of these therapies.15 The two main clinical phenotypes are a typical length-dependent sensory axonal neuropathy and an acute demyelinating polyradiculoneuropathy mimicking Guillain-Barré syndrome. Due to the small number of reported cases, the optimal treatment regimen is unknown, although we have seen complete recovery with high-dose corticosteroids alone.
Management of chemotherapy induced peripheral neuropathy
Accurate and confident characterisation by a neurologist is essential to therapeutic decision-making by the oncologist or haematologist. Patients with chemotherapy-induced peripheral neuropathy due to traditional chemotherapies can be reassured that most will improve over months, though a significant proportion have some residual and long-lasting symptoms. Except for immune checkpoint inhibitors and BRAF and MEK inhibitors, there are no disease-modifying treatments, though neuropathic pain medications may be useful symptomatically, with randomised controlled trial evidence for duloxetine in painful chemotherapy-induced peripheral neuropathy.16
Other medications
Table 3 summarises other potentially neurotoxic medications, discussed below.
Medications associated with peripheral neuropathy
Sensory predominant
Pyridoxine
Pyridoxine (vitamin B6) is found in meat, eggs, grains and green vegetables, and the recommended daily intake is up to 2 mg. It is also popular as a dietary health supplement. Deficiency is rare in people with a normal diet but develops in malnutrition through starvation, alcoholism or anorexia, or in times of increased metabolic demand such as pregnancy and systemic illness. Isoniazid and levodopa/carbidopa intestinal gel can also lead to deficiency through altered metabolism.
While pyridoxine deficiency causes an ascending sensory axonal neuropathy, pyridoxine toxicity typically results in a sensory ganglionopathy, with sensory loss in the limbs and sometimes the face and trunk and an ataxic gait.17 Histopathological studies have shown selective degeneration of sensory neurones of the dorsal root ganglia and their central and peripheral processes and sensory distal axonopathy; however, the mechanism for toxicity is unknown.18 Most multivitamins contain 1.7–2.0 mg of pyridoxine, and B-complex supplements often contain 2–6 mg.19 Neuropathic symptoms usually arise from acute ingestion of extremely high doses (eg, 180 g) or chronic ingestion of widely available dedicated ‘high strength’ vitamin B6 supplements (≥50 mg daily).19 The cut-off plasma concentrations for pyridoxine toxicity have not been clearly defined; mildly raised concentrations (2–3 times the upper limit of normal) are common in people taking multivitamin or B-complex supplements and are usually not clinically relevant. In the setting of a raised pyridoxine concentration, we advise patients to stop vitamin B6 supplements; however, a neuropathy should not be attributed to pyridoxine toxicity unless there is clear improvement or stabilisation after stopping the supplement.
Antituberculous drugs
Isoniazid is well known to cause a sensory predominant neuropathy, which is secondary to acquired pyridoxine (vitamin B6) deficiency.20 This is now largely prevented by routine pyridoxine supplementation. Optic neuropathy (commonly associated with neuropathies due to nutritional deficiencies) and CNS manifestations may also occur. Ethambutol more commonly causes optic than peripheral neuropathy; however, there have been a few cases of axonal sensory neuropathy described, with improvement after stopping the drug.21
Triazole antifungals
Neuropathy in patients taking triazole antifungals can be secondary to CYP3A4 inhibition, increasing serum concentrations of other neurotoxic drugs (vinca alkaloids or calcineurin inhibitors). However, a usually mild predominantly sensory neuropathy may occur in people taking itraconazole, voriconazole or posaconazole without other neurotoxic agents, occurring in around 10% after an average of 4 months, and usually resolving with stopping the drug.22
Phenytoin
The most common neuropathic deficit from chronic phenytoin use is asymptomatic nerve conduction abnormalities, occasionally with symptoms of a mild sensory predominant length-dependent neuropathy. Phenytoin-associated cerebellar dysfunction is much more frequent. There are also reports of more clinically significant acute and chronic neuropathies with significant weakness, often associated with high doses and/or plasma concentrations in the absence of cerebellar signs, and generally reversing on stopping the drug.23
Antiretrovirals
An axonal distal sensory predominant polyneuropathy develops in 10%–35% of people with HIV and is more common and severe with increasing immunosuppression.24 Some of the older nucleoside reverse transcriptase inhibitors (stavudine, didanosine and zalcitabine) can cause a clinically similar neuropathy, although these drugs are no longer available in the UK and many other countries. A neuropathy in someone with HIV in most developed countries is now unlikely to be caused by their antiretroviral treatment.
Fluoroquinolones (eg, ciprofloxacin, norfloxacin)
While these antibiotics may increase the risk of neuropathy, this appears to be extremely rare, with the estimated number needed to harm approximately 150 000 with a 10-day course.25 Most reports are of predominantly sensory symptoms26; however, there is little detailed information on the clinical features and purported pathogenic mechanism in the literature, and clinicians should be cautious when attributing causation.
Leflunomide
Leflunomide, used in rheumatoid arthritis, can cause a length-dependent sensory or sensorimotor neuropathy, usually after months of treatment; this may improve if the drug is stopped soon after symptom onset.27
Colchicine and chloroquine
Colchicine and chloroquine are much more commonly associated with myopathy than neuropathy; however, many patients with myopathy also have a mild sensory neuropathy, particularly with chronic use.28 29
Drug-induced neuropathies that may have significant distal motor involvement
Dapsone
Dapsone can cause a motor predominant neuropathy, mainly with prolonged treatment (months to years).30 Most cases improve markedly with stopping the drug. Dapsone is used for leprosy (the most common cause of neuropathy in low-income countries). However, leprous neuropathy is typically a multiple mononeuropathy and includes sensory involvement, making differentiation straightforward.
Nitrofurantoin
Nitrofurantoin has been associated with a length-dependent axonal neuropathy of rapid onset, often with significant motor involvement clinically.31 There is typically distal weakness and wasting, and sometimes pain, meaning it can be confused with Guillain-Barré syndrome and vasculitis. The neuropathy appears to be idiosyncratic and not related to dose, and occasionally occurs with short courses, but is probably a rare side effect.
Disulfiram
Disulfiram, used to assist with abstinence in overusers of alcohol, can cause a sensorimotor axonal neuropathy, often with both muscle weakness and sensory symptoms.32 The symptoms often improve markedly once disulfiram is stopped, and this is maintained even if alcohol consumption resumes.
Amiodarone
Amiodarone has several neurological side effects, including tremor, cerebellar ataxia, optic neuropathy, peripheral neuropathy and myopathy. The neuropathy develops over weeks to months, is typically distal predominant and can significantly involve both motor and large sensory fibres, giving both distal weakness and sensory ataxia.33 Nerve conduction study findings vary from predominant conduction velocity slowing to predominant reduction in motor and sensory amplitudes. Similarly, nerve biopsies may show segmental demyelination or predominant axonal loss. The biopsy may also show crystalline intralysosomal inclusions, which may be involved in the pathogenesis.
Painful
Linezolid
The oxazolidinone antibiotic linezolid has been associated with an often painful predominantly sensory axonal neuropathy, mainly with prolonged use over several months.34 Optic neuropathy may also occur and is more likely to improve than the peripheral neuropathy.
Metronidazole
Metronidazole can cause a distal sensory neuropathy, with patients often reporting burning pain. It tends to occur with prolonged use (≥4 weeks) and sometimes high-dose treatment, but appears to be very rare with the short courses commonly used for infections.35 The neuropathy is sometimes accompanied by other features of neurological toxicity (eg, ataxia, encephalopathy), and in most cases reverses with stopping the drug (case 1).
Case 1
A 60-year-old man reported numbness, aching pain and weakness in his legs for 2 months. He had a history of primary sclerosing cholangitis for 10 years, ulcerative colitis and hypertension. Medications included prednisolone, azathioprine, bumetanide, metronidazole, ursodeoxycholic acid, omeprazole, candesartan and amlodipine. The metronidazole (400 mg three times daily) had been started 6 months earlier due to recurrent episodes of cholangitis and worsening liver function tests. On examination, there was weakness of toe extension bilaterally, absent ankle jerks, reduced pinprick sensation to the upper calves and reduced vibration sensation to the right knee and left costal margin. Nerve conduction studies showed a length-dependent sensorimotor axonal polyneuropathy, with absent lower limb and reduced upper limb sensory amplitudes, and a reduced peroneal motor amplitude to extensor digitorum brevis. A neuropathy screen including extensive testing for nutritional deficiencies was unrevealing. Metronidazole was considered a likely cause of the neuropathy and was stopped with the agreement of the patient’s gastroenterologist. At reassessment 3 months later, the pain had resolved and the sensory loss had receded to the ankles. Repeat nerve conduction studies a further 2 months later showed normal motor studies and upper limb sensory amplitudes, with absent lower limb sensory responses.
Analysis:
Metronidazole-induced neuropathy usually occurs with very prolonged courses (≥4 weeks). Like many other toxic neuropathies, improvement can occur with stopping the offending agent.
Note that several other drug causes listed may sometimes have pain as a significant feature.
Acute/subacute/chronic onset polyradiculoneuropathy
Tumour necrosis factor (TNF) inhibitors
Anti-TNF drugs are used in autoimmune conditions including rheumatoid arthritis, sarcoidosis and inflammatory bowel disease. In addition to known immune neurological side effects (eg, CNS demyelination), they are rarely associated with acute or subacute onset neuropathies, particularly polyradiculoneuropathy (Guillain-Barré syndrome or CIDP-like presentation).36 In some cases the drug may have led to infection through immunosuppression; however, an immune mechanism remains possible. Mononeuropathy multiplex (vasculitis-like) presentations may also occur, although it may be difficult to attribute causation to the drug if the underlying condition is an inflammatory arthritis (which can cause peripheral nerve vasculitis). Patients may improve when treatment is stopped; however, some patients require immunomodulation (intravenous immunoglobulin or plasma exchange).
Calcineurin inhibitors
Tacrolimus and ciclosporin are well known to cause neurological adverse effects, including tremor, seizures, optic neuropathy and posterior reversible encephalopathy syndrome. Post-transplant patients taking calcineurin inhibitors have a higher incidence of neuropathic symptoms and signs than those receiving other medications.37 Guillain-Barré syndrome and CIDP-like presentations may also occur, almost exclusively in post-transplant patients, and it is difficult to disentangle the causation with complex interventions such as these.38
Interferon-alpha
Interferon-alpha used for viral hepatitis has been associated with polyradiculoneuropathy, particularly CIDP.39 Most reported cases required treatment with intravenous immunoglobulin, plasma exchange or corticosteroids in addition to stopping the drug; however, most did not need long-term treatment.
Recreational drugs
Alcohol
Nearly half of chronically alcohol-dependent people are affected by peripheral neuropathy.40 Patients who chronically overuse alcohol develop an insidious, length-dependent, often painful sensory axonal neuropathy that predominantly affects the lower limbs. While excessive alcohol impairs thiamine absorption, storage and metabolism, it has been shown that alcohol-related neuropathy is distinct from the neuropathy due to beriberi (thiamine deficiency neuropathy). Well-nourished alcoholics can develop neuropathy despite supplementation of alcohol with thiamine and pyridoxine.41 A Japanese study compared patients with pure alcohol-related neuropathy, patients with beriberi and patients with concurrent alcohol-related neuropathy and thiamine deficiency.42 Those with pure thiamine deficiency developed a rapidly progressive motor-predominant axonopathy, with loss of large fibres and extensive subperineural oedema on nerve biopsy. In contrast, those with solely defined alcohol-related neuropathy had a slower progressive onset with predominant sensory involvement, and biopsy findings of small fibre loss. Despite this distinction, in practice the neuropathy in many alcoholics is multifactorial, with direct toxic effects and nutritional deficiencies contributing.
Nitrous oxide
In addition to its clinical use as a weak anaesthetic, nitrous oxide (N2O) is also used recreationally, particularly among teenagers and young adults. It is often obtained as canisters (‘whippits’) to use for making whipped cream (figure 2) and is inhaled via a balloon. Data for 2018/2019 found that 2.3% of adults aged 16–59 in the UK (around 763 000 people) had used nitrous oxide in the last 12 months.43 Over recent years, the increased availability of extra-large cannisters intended for industrial use has increased the frequency of presentations due to nitrous oxide toxicity.

{kind=link}
{kind=link}
Empty nitrous oxide canisters in a London residential street, February 2020. From: Sobczyńska-Malefora et al.59 Reproduced under the terms of the Creative Commons BY licence.
Nitrous oxide oxidises cobalt ions in vitamin B12 (cobalamin), rendering methylcobalamin (a physiologically active form of B12) inactive. Interference with B12-dependent pathways reduces conversion of homocysteine to methionine, causing damage to the dorsal columns of the spinal cord and to the peripheral nervous system.44 Toxicity occurs mainly in recreational users, but can also occur with repeated high doses given in a clinical setting, especially in those with low or borderline plasma B12 concentrations. Inadvertent replacement of B12 by drinking yeast-containing beer in parallel protects against nitrous oxide related toxicity, influencing the demographic of those presenting.
Chronic or excessive exposure to nitrous oxide may result in subacute combined degeneration of the spinal cord, characterised by predominant demyelination of the dorsal columns (leading to sensory ataxia) with varying degrees of motor involvement. The neuropathy due to B12 deficiency is usually length-dependent and is predominantly sensory.45 However, nitrous oxide toxicity may also cause an acute or subacute onset distal and motor predominant axonal neuropathy (case 2).46
Case 2
A 24-year-old man reported difficulty walking and leg numbness, which had acutely worsened over 1 week. He had a 3-year history of similar but less severe lower limb symptoms and a 2-year history of bladder urgency. He admitted to using nitrous oxide for the past 5 years and was using up to 60 bottles per day at presentation. On examination, he walked with a bilateral foot drop gait. Upper limb examination was normal. Lower limb strength was normal proximally, but with severe weakness of ankle dorsiflexion and eversion and mild weakness of plantarflexion and inversion. Lower limb reflexes were absent and the right plantar was upgoing. He had reduced pinprick perception to the ankles with normal vibration sense and proprioception. Plasma B12 was normal (218 pg/mL; normal 197–771) but plasma homocysteine was elevated (43.1 μmol/L, normal 5–12). MR scan of the spine was not available. On nerve conduction studies, lower limb motor amplitudes were severely attenuated and lower limb sensory amplitudes were present, though reduced for age (right 6.2 μV, left 8.0 μV). He was diagnosed with complications of nitrous oxide abuse and started on daily vitamin B12 injections. He reported some improvement but continued to use nitrous oxide (24 bottles per day) and bladder symptoms persisted.
Analysis:
This was a motor-predominant neuropathy due to nitrous oxide use, with minimal spasticity (upgoing right plantar and bladder urgency) that likely related to concurrent myelopathy. B12 concentrations are commonly normal in nitrous oxide toxicity, thus homocysteine and/or methylmalonic acid should also be tested.
Plasma B12 concentrations may be normal in nitrous oxide toxicity, and thus it is essential to also check homocysteine and methylmalonic acid concentrations, which are typically markedly elevated.46 Treatment relies on B12 replacement, stopping nitrous oxide use and neurorehabilitation. High-dose intramuscular B12 loading is often given (eg, 1 mg every 2 days for 11 doses), followed by long-term replacement with 1 mg every 2–3 months. Neurological recovery can often be slow and may be incomplete.46
Heavy metals
Lead
Lead toxicity is now rare, with significant exposure mainly confined to specific industries (eg, smelting and alloying, lead battery manufacture).47 In the UK, the Health and Safety Executive carefully monitors these industries, and workers with potential significant exposure have regular monitoring of blood lead concentrations. Despite this, occasional cases of lead toxicity still occur in industry in less closely monitored countries, and some lead-containing materials (eg, solder) are still obtainable for home use. Furthermore, some traditional medicines contain or are contaminated by lead or other heavy metals.
In contrast to most toxic neuropathies, lead causes a subacute onset, predominantly motor neuropathy, with early and preferential involvement of the wrist and finger extensors, and sometimes asymmetry.48 A chronic sensory neuropathy with prolonged exposure is less common. Lead toxicity may be accompanied by cognitive and behavioural changes. Nearly all patients develop a microcytic anaemia with basophilic stippling of red cells, and most also have other organ involvement, particularly gastrointestinal (abdominal pain, constipation) and renal (proteinuria, renal impairment). Lead neuropathy is treated with chelation therapy, and generally has a good prognosis once the exposure is removed.48
Mercury
Mercury exists in elemental, inorganic and organic forms, with exposure to elemental and inorganic mercury occurring mainly in industry (eg, manufacture of thermometers, chloralkali industry), and exposure to organic mercury due to ingestion of contaminated seafood. Organic mercury can cause CNS toxicity, whereas chronic elemental or inorganic mercury poisoning causes a length-dependent predominantly sensory neuropathy with ataxia, often accompanied by stomatitis, behavioural changes, a postural tremor and renal impairment.49 Diagnosis may be achieved by testing 24-hour urine mercury concentration, which mainly reflects exposure to elemental and inorganic mercury. Improvement normally occurs once exposure to mercury is removed, though chelation is usually also recommended.
Arsenic
Inorganic arsenic toxicity occurs in the settings of contaminated drinking water, agricultural (herbicides, pesticides) and occupational exposure (mining, smelting) and events with homicidal or suicidal intent.50 It is also an ingredient in some Chinese and Indian traditional medicines. Acute poisoning causes a severe gastrointestinal illness (vomiting, abdominal pain, diarrhoea), which may be followed several weeks later by a rapid onset polyradiculoneuropathy similar to Guillain-Barré syndrome.50 Chronic exposure may cause a mild sensorimotor axonal neuropathy, often with concurrent gastrointestinal symptoms, hyperpigmentation and keratosis, Mees’ lines and anaemia. Organic arsenic from seafood consumption can be detected in urinary screens and is not known to be harmful. Patients should thus be advised to refrain from consuming seafood for 5 days before sampling. Arsenic trioxide is used to treat acute promyelocytic leukaemia, although significant neuropathy is uncommon at standard doses.
Thallium
Thallium was historically an ingredient in rodenticides and is used in several industries, although occupational exposure is rare. It has also been used as an agent of homicidal intent. Thallium toxicity typically manifests hours after ingestion with gastrointestinal symptoms (nausea, vomiting, abdominal pain) and a painful sensorimotor neuropathy, which can range from mild distal symptoms to an acute and severe illness similar to Guillain-Barré syndrome.49 Autonomic and neuropsychiatric symptoms may appear, and patients usually develop alopecia several weeks following the onset of illness.
Cadmium
Cadmium has a very long half-life (15–20 years), and those exposed in industry (eg, battery manufacture, smelting) may develop late toxicity. In addition to causing renal and pulmonary toxicity, anosmia and neurobehavioural effects, cadmium exposure has been associated with sensory neuropathic symptoms and has been found to accumulate in dorsal root ganglia in animal models.51
Diagnostic testing for heavy metal poisoning
Measurement of blood and urine concentrations of heavy metals can be informative in cases of recent exposure. On the other hand, if the exposure occurred weeks before blood and urine sampling, heavy metal concentrations may be normal. Quantification of heavy metal content in hair can be used to detect historic heavy metal intoxication but is usually available only via forensic rather than healthcare laboratories.
Treatment of heavy metal poisoning
Supportive care and prevention of further exposure remain cornerstones of treatment for most cases of heavy metal poisoning. Chelation agents may be recommended for significant lead, mercury or arsenic toxicity, with specific guidelines available from the clinical toxicology database of the National Poisons Information Service (TOXBASE, www.toxbase.org). Thallium toxicity can be treated with Prussian blue.
Ciguatera and other biological toxins
Ciguatoxin is synthesised from benthic (bottom dwelling) microorganisms that grow in association with algae in reef areas. It accumulates in the tissues of small reef fish that consume them, which in turn are eaten by larger fish. Ciguatoxin remains present in frozen fish and can persist for up to 6 months. Contaminated fish have normal appearance, aroma and taste.52 Endemic areas include the Caribbean, south-east Asia, eastern Australia and the Pacific. Ciguatoxin binds to and opens voltage-dependent sodium channels, leading to depolarisation and spontaneous or repetitive firing of action potentials.52 Most affected individuals develop symptoms within hours of ingesting the toxic fish. The toxidrome begins with acute gastrointestinal symptoms followed by perioral paraesthesia and dysaesthesia spreading to the extremities. A distinctive symptom is paradoxical temperature reversal, whereby cold objects feel intensely hot (and vice versa). Nerve conduction studies may show slowed sensory and motor conduction velocities and prolonged F-waves,53 or there may be only small fibre dysfunction.52 Treatment is largely supportive, although in severe cases there may be a role for mannitol. The recovery takes days to weeks. People previously affected are more susceptible to developing symptoms if re-exposed.
Saxitoxin, found in contaminated shellfish, and tetrodotoxin, produced by the fugu (puffer fish), are both potent sodium channel blockers. Fugu is considered a Japanese delicacy and requires meticulous preparation. Consumption of saxitoxin or tetrodotoxin can cause rapid onset of perioral and limb paraesthesia, severe weakness and even death.54 There is no antidote for either of these toxins, but immediate intervention with cardiorespiratory monitoring leads to recovery in most cases.
Industrial agents
Acrylamide
Acrylamide is used in the dye, paper, plastic, adhesive and grout manufacture industries and in water processing. It is toxic to nerve terminals and Purkinje neurones. Acrylamide probably inhibits kinesin-based fast axonal transport, alters neurotransmitter levels and directly inhibits neurotransmission.55 Chronic exposure to the acrylamide monomer leads to skin irritation and hyperhidrosis but also a length-dependent pansensory-motor polyneuropathy, with prominent sensory ataxia.
n -hexane and ‘glue-sniffing’
n-hexane is a six-carbon chain ring solvent used in the industrial setting for glue and textile manufacture and also recreationally as ‘glue-sniffing’. It is neurotoxic and causes a distal sensorimotor neuropathy, with progressive sensory symptoms and weakness. Onset can be acute or insidious depending on the dose. Nerve conduction studies usually show axonal features, though there may be significant conduction slowing, conduction block and temporal dispersion in acute cases and with high doses.49 Increased concentration of hippuric acid in blood or urine may indicate recent ‘glue-sniffing’.56
Organophosphates
Organophosphorus compounds collectively inhibit cholinesterase enzymes causing muscarinic overactivity and acute neuromuscular blockade. Worldwide, they are commonly used as pesticides and insecticides in the agricultural industry, but exposure may also arise from suicide attempts or poisonings. Transdermal, respiratory or gastrointestinal absorption can result in toxicity, which in the acute phase presents as cholinergic crisis and classic ‘SLUDGE’ (salivation, lacrimation, urination, defaecation, gastrointestinal illness, emesis) syndrome, and its later effects include neuropathy. The neuropathy arises around 3 weeks after exposure and includes distal numbness, paraesthesiae, cramping and weakness with bilateral foot drop.57 Sensory disturbance is usually mild and may be absent. Nerve conduction studies show reduced motor amplitudes without substantial conduction slowing. While atropine can be an antidote for acute toxicity, neuropathy management is symptomatic.
Other industrial agents that can cause neuropathy include diethylene glycol (acute Guillain-Barré syndrome-like presentation), allyl chloride, carbon disulphide, ethylene oxide and dimethylaminopropionitrile. Except for severe cases, the prognosis in most neuropathies due to industrial agents is relatively good once the toxin is removed.
Conclusions
Toxic neuropathies comprise a heterogeneous group of conditions that can cause significant morbidity and impairments. Since many toxic neuropathies improve with removal of the offending agent, it is essential that neurologists can recognise them. A careful history and examination, with attention to the temporal course and the neuropathic and systemic features, will allow the diagnosis in most cases.
Key points
Clinical assessment of a possible toxic neuropathy should focus on noting the temporal association between exposure and symptoms, and identifying the neuropathy phenotype and non-neuropathic symptoms and signs.
The most common causes of toxic neuropathy worldwide are alcohol and medications, particularly chemotherapeutic agents; both traditional and newer antineoplastic drugs may cause neuropathy.
Removal of the responsible toxin in many cases leads to improvement or prevents further progression; however, a lack of response should prompt a search for alternative causes.
While biochemical tests are useful, they can aid but cannot replace the clinical history and examination.
Further reading
Staff, N. P., Grisold, A., Grisold, W., & Windebank, A. J. Chemotherapy-induced peripheral neuropathy: A current review. Ann neurol 2017, 81(6), 772–781.
Park SB, Goldstein D, Krishnan AV, et al. Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J Clin. 2013;63(6):419–37.
Little AA, Albers JW. Clinical description of toxic neuropathies. Handb Clin Neurol. 2015;131:253–96.
Achaibar KC, Moore S, Bain PG. Ciguatera poisioning. Pract Neurol 2007;7 (5):316–322.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Twitter @duncansmyth, @mike_the_nerve
Contributors DS and MPL were involved in the design and conception of the paper. DS and CK wrote the first draft. DS, CK, ASC, AMR and MPL edited, critically revised and approved the final manuscript for submission.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests DS is supported by the New Zealand Neurological Foundation. CK is supported by a UCL Queen Square Institute of Neurology and Cleveland Clinic London MPhil/PhD Neuroscience fellowship. ASC, AMR and MPL are supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. DS, CK, ASC, AMR and MPL received no funding or sponsorship for this commissioned paper. ASC has received honoraria from CSL, Grifols, Akcea, BMS, BeiGene and Lupin for educational talks and advisory input. MPL has provided consultancy for UCB Pharma, CSL Behring and Polyneuron. He was the principal investigator on trials with Polyneuron and UCB Pharma for which his institution receives investigator fees. He has been on the data safety monitoring board for Octapharma, IoC trial, AstraZeneca Pharmaceuticals.
Provenance and peer review Commissioned; externally reviewed by Tim Lavin, Manchester, UK.
Other content recommended for you
- Peripheral neuropathy: pattern recognition for the pragmatist
- Peripheral neuropathy: pattern recognition for the pragmatist
- Peripheral neuropathy in complex inherited diseases: an approach to diagnosis
- Ultrasound scanning in the diagnosis of peripheral neuropathies
- Iatrogenic immune-mediated neuropathies: diagnostic, epidemiological and mechanistic uncertainties for causality and implications for clinical practice
- Peripheral neuropathy
- Diagnostic investigation of patients with chronic polyneuropathy: evaluation of a clinical guideline
- Pragmatic guide to peripheral nerve disease and the role of clinical biomarkers
- Neuropathy associated with gluten sensitivity
- Ultrasensitive assay technology and fluid biomarkers for the evaluation of peripheral nerve disease