Article Text

Abstract
Posterior cortical atrophy (PCA) is a neurodegenerative syndrome dominated by deterioration of higher visual function (particularly visuospatial and visuoperceptual abilities). It is most commonly due to Alzheimer's disease pathology, but may also be caused by dementia with Lewy bodies, corticobasal degeneration or Creutzfeldt-Jakob disease. Patients often present to optometrists, ophthalmologists and/or neurologists with non-specific visual complaints, and unless clinicians seek the specific symptoms and signs of PCA (beyond that of the ‘standard’ neurological examination), this infrequent disorder is easily missed, delaying its diagnosis and treatment. We review the clinical features of PCA, focusing on its visual manifestations, to help neurologists recognise this important syndrome.
- Posterior Cortical Atrophy
- Balint's syndrome
- Simultanagnosia
- Alzheimer's disease
- Neuro-ophthalmology
Statistics from Altmetric.com
Introduction
Posterior cortical atrophy (PCA), typically considered a focal variant of Alzheimer's disease, is a neurodegenerative syndrome dominated by deterioration of visuospatial, visuoperceptual, numeracy and literacy abilities. The erosion of these skills reflects the pathological atrophy of the primary visual cortex, as well as the dorsal (occipitoparietal) and ventral (occipitotemporal) visual streams. Patients typically report vague or even bizarre visual symptoms; frequently, the ‘standard’ ophthalmic and neurological examinations are normal. This confusing clinical scenario often causes delay in the diagnosis of PCA, with delay in starting appropriate treatments. We review the features of PCA, focusing on its visual manifestations (box 1).
Case vignette: an ‘iconic’ presentation
A right-handed 60-year-old woman, previously healthy, presented to the neuro-ophthalmology clinic with vision difficulties. She worked as a landscape architect having previously retired as a social science researcher, and had a PhD in regression analysis. Eighteen months before this assessment, she had noticed difficulty reading. Most striking was her description of visual confusion when multiple words were present on a single page. Attributing it to presbyopia, she tried enlarging the font on her Kindle device but this only worsened the problem.
An optometrist and ophthalmologist found no abnormalities. A neurologist also found nothing wrong and interpreted her brain MRI as normal. She continued to complain emphatically about her reading problems, prompting referral to our institution's low vision centre. She confirmed a peculiar facet of her reading difficulties: that her reading ability improved by decreasing font and column sizes, rather than by enlarging them. Despite her self-described sophistication with computer technologies, she reported significant difficulty using her computer. Specifically, she could not navigate the computer screen as she had difficulty seeing all the displayed items. Furthermore, she found the email interface (the programme she had used without difficulty for some time) confusing to the extent that she could no longer discern meaningful relationships among the various icons. She had particular difficulty acknowledging and differentiating among various textures and patterns, although this had been an important reason for her success as a landscape architect.
From the safety perspective, she noted progressively worsening ability to navigate kerbs and steps, particularly those in highly patterned materials. There was no leg weakness or sense of imbalance. She found difficulty driving in unfamiliar surroundings, and eventually became disorientated in familiar environments. Eventually, she and her husband noticed problems with short-term memory.
On neuro-ophthalmic examination, she had remarkable difficulty reading the Hardy-Rand-Rittler and Ishihara pseudoisochromatic charts. While she could not perceive the figures, or finger-trace them, she readily identified the colours of the individual circles that made up the plates. Visual acuity was 6/9 (20/30) bilaterally, improving to 6/6 (20/20) with pinhole correction. She had difficulty reading a line of letters, but could manage single letters. She perceived the 6/9 (20/30) line of the Snellen chart more easily than the 6/240 (20/800) line. Stereoscopic vision was normal. Confrontational visual field testing was inconsistent but appeared normal. Goldmann kinetic perimetry showed right hemifield paracentral scotomas (figure 1). She had a concomitant esophoria, and full extraocular movements. Vertical and rightward saccades were hypometric. Leftward smooth pursuit eye movements were saccadic, and optokinetic quick-phases to the right were impaired. Pupillary reflexes, slit-lamp examination and dilated funduscopy were normal.
Although alert and orientated to time, place and person, she had striking difficulty in recalling her age, but no problem remembering her date of birth. Her naming, comprehension, repetition, writing and immediate recall were intact. She recalled two of three objects at 5 min. She could not perform serial 7s or any calculations, but could spell ‘WORLD’ backwards. She could write a sentence, albeit slowly, but could not copy intersecting pentagons. Her reading was laboriously slow but intact; her letter-by-letter reading was better than whole-word reading.
She described the Boston cookie-theft picture in a piecemeal fashion; for example, she identified ‘a boy leaning backwards’ and ‘a lady chef’. With Navon figures, she easily identified the smaller letters but failed to appreciate the global figures. She could not imitate interlocking finger figures (a rapid bedside screening-test for parietal lobe dysfunction). While she drew a clock face adequately, but could not ‘set’ time, suggesting greater impairment of the left parietal lobe. There was no ideomotor apraxia, dressing apraxia, optic ataxia, ocular apraxia, finger agnosia, right-left disorientation, prosopagnosia, achromatopsia, agraphesthesia, astereognosis, hemi-inattention, parkinsonism, alien limb, myoclonus or frontal release signs. The remaining neurological examination was normal.
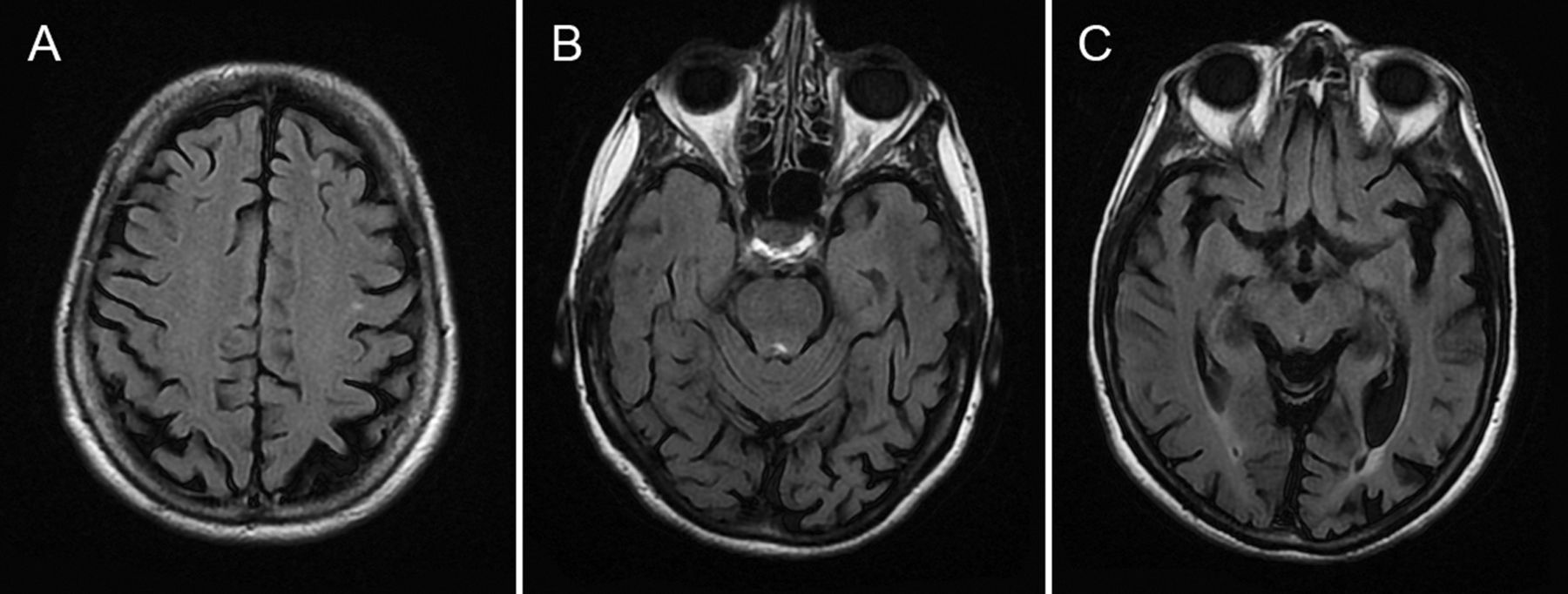
We made a clinical diagnosis of posterior cortical atrophy, supported by the brain MRI (figure 2).
Clinical features
The age of onset of PCA is earlier than that of amnestic Alzheimer’s disease, with most studies reporting an age of onset from the mid-50s to the early 60s.1 The clinical picture of PCA is dominated by visuoperceptual and visuospatial impairments, typically in the context of astonishingly normal ophthalmological findings. Specifically, the salient features of PCA include Balint's syndrome (simultanagnosia, optic ataxia and ocular apraxia), Gerstmann's syndrome (agraphia, acalculia, right-left confusion and finger agnosia), visual agnosia, alexia with or without agraphia, and environmental agnosia (leading to topographical disorientation). Additionally, there may be alexia, anomia, environmental disorientation, apraxia, hemi-inattention, prosopagnosia and transcortical sensory aphasia.2––7 A complete Balint's syndrome and/or Gerstmann's syndrome is rare at first presentation. Instead, it is more common to have components of either syndrome (usually simultanagnosia in Balint's syndrome and acalculia in Gerstmann's syndrome).6 ,8––10 As the neurodegenerative processes spread, causing the patient to deteriorate, other cardinal features of these syndromes emerge. Some authors subdivide PCA into two forms—a biparietal form that predominantly affects the dorsal ‘where’ visual pathway (subsequently leading to visuospatial deficits) and an occipitotemporal form that mainly affects the ventral ‘what’ visual pathway (resulting in problems of object recognition and identification) (see online supplementary file reference S2). However, others argue that such a dichotomy may only be relevant in the early phases of the disorder10 (see online supplementary file reference S3).
In contrast to amnestic Alzheimer's disease, patients with PCA typically show preservation of memory, insight, language skills and judgment until late in the clinical course, when the clinical features of PCA and Alzheimer's disease overlap.2 ,7 ,8 ,11 As such, prominent cortical visual dysfunction with relatively normal insight, verbal memory and fluency in an older adult should always raise the suspicion for PCA. Additionally, unlike amnestic Alzheimer's disease, patients with PCA are often acutely aware of their visual impairment since their insight and cognitive abilities remain intact (notwithstanding that these visual findings are often missed or dismissed by clinicians); as a result, patients often develop reactive anxiety and depression.1 ,7 ,8 The presenile onset of the disease, its rarity, its variable clinical features (especially when combined with a normal ocular examination), as well as the presence of a mood disorder, may cause even the most experienced neurologist to misdiagnose these patients as ‘non-organic’, ‘anxious’ or ‘functional’.
Visual manifestations
The visuoperceptual and visuospatial manifestations of PCA often compel patients to seek medical attention. Early complaints are non-specific and often relate to visual ‘blurriness’, and/or glare sensitivity.1 These complaints often prompt them to seek optometric and ophthalmological evaluations; these examinations are almost always normal, and may even prompt unnecessary cataract surgery.4 ,5 ,12 ,13 Many patients are subsequently referred to neurologists to determine the cause of their visual complaints.
The most common and disabling impairment that patients with PCA first notice is reading difficulty.2 ,3 ,10 ,11 ,13––18 Several processes conspire to impair reading ability, including visual disorientation (failure to track a line of text, or losing one’s place on the page),1 ,2 ,16 ,19 simultanagnosia,1 ,15 ,20 visual crowding (a form of masking where neighbouring stimuli impede the identification but not detection of a target),1 ,20 reverse-size phenomenon, alexia,1 ,2 ,15 ,21 and/or ocular apraxia (failure to generate saccades to move from word to word). Additionally, patients with PCA may report difficulty reading an analogue clock face—a possible manifestation of simultanagnosia.
Similarly, patients may be unable to perceive textures and patterns (as in our case vignette). In most, this affects their ability to walk on patterned surfaces; however, in those whose occupation relates to art and design, this may be particularly bothersome. The difficulty in perceiving patterns/textures may result from simultanagnosia, visual disorientation and/or saccadic dysfunction. Disruption in other neural processes may also contribute, including impaired depth perception,19 length/orientation discrimination,22 contrast sensitivity23 and complex visual attention.24
Homonymous visual field deficits, in particular homonymous hemianopia or quadrantanopia, are frequently present but underdiagnosed, probably because patients’ cognitive deficits hamper accurate perimetric examination.1 ,13 ,17 Not uncommonly, there may be incongruous and variable deficits8; such findings may lead some clinicians to dismiss the patients’ visual complaints as ‘non-organic’. In the more typical amnestic form of Alzheimer's disease, visual field perimetry not uncommonly shows bilateral inferior field constriction.12 Thus, inferior field deficits in a patient who otherwise has features characteristic of amnestic Alzheimer's disease does not necessarily have PCA.
Simultanagnosia
No discussion of PCA is complete without discussing its most striking visual manifestation—simultanagnosia. The human visual system, due to its limited processing capacity, manages the myriad of individual elements in any scene by categorising it based on gestalt (ie, seeing the forest before the trees); this is known as the global precedence effect,25 and helps us make sense of the rich visual scenes that confront us daily. Simultanagnosia is a neuropsychological disorder characterised by the inability to synthesise the overall meaning of a visual scene despite being able to recognise its individual elements at any given time (ie, seeing the trees but not the forest). This debilitating condition is the consequence of lesions affecting both parieto-occipital junctions, particularly Brodmann area 7 (also responsible for driving saccades to novel stimuli) and the superior occipital cortex.2 ,26––29 Patients with simultanagnosia cannot integrate individual objects to create a coherent visual representation of the world, and as such, can perceive only one object at a time. In severe cases, they perceive only constituent parts of a larger object (sometimes causing ‘partonomic’ errors, mistaking parts of objects as whole objects).6 ,29 Thus, the patient's visual world becomes ‘unglued’—scenes and objects are perceived piecemeal, rather than in a global manner.29 This abnormal direction of attention towards smaller, local elements of a scene at the expense of global features is termed ‘local capture’.27 ,30 Patients with simultanagnosia are often functionally blind, with severely impaired activities of daily living.29
Patients’ descriptions of simultanagnosia can be vivid and arresting. For example, Coslett and Saffran26 described a patient who “reported watching a movie in which, after a heated argument, she noted to her surprise and consternation that the character she had been watching was suddenly sent reeling across the room, apparently as a consequence of a punch thrown by a character she had never seen.” In more dramatic cases, patients report spontaneous disappearance of objects from view, despite eye movement recordings showing that they were fixating on those objects (‘looking but not seeing’).31 In early PCA, however, simultanagnosia may be subtle and impair reading abilities and activities of daily living. Some patients may tend to lose items, and attribute this to misplacing them; their family members may report that these patients seem unable to see or locate objects that are lying in plain sight—an important diagnostic clue. Like our case vignette, reading becomes problematic, and patients may discover (much to the patient's and clinician's bewilderment) that larger fonts are more difficult to perceive than smaller fonts; this ‘reverse-size’ phenomenon probably results from a narrower window of visual attention (discussed later).
A highly conspicuous sign of simultanagnosia is the inability to read pseudoisochromatic plates, despite intact colour perception. The Ishihara and Hardy-Rand-Rittler pseudoisochromatic plates are pigment-based colour vision tests designed to evaluate colour vision impairment, especially congenital deficiencies. The patient has to combine circles of similar colour visually into a number/figure that stands out from the background. Thus, these tests require the ability to see various colours and the ability to combine the dots into a whole figure. While patients with simultanagnosia can perceive colours, they cannot combine the multiple local elements and synthesise the whole figure,28 analogous to their inability to perceive the global precept of Navon letters. Many patients with PCA show this discordance between colour identification and Ishihara/ Hardy-Rand-Rittler pseudoisochromatic chart performance.15 ,18 ,21 ,28 Similarly, patients with simultanagnosia cannot perceive the degraded letters (presented on a background of random black pattern stimuli) of the Visual Object and Space Perception Battery. The Boston cookie-theft picture is also an effective bedside screening tool for simultanagnosia. Like our case vignette, patients often see ‘a boy leaning backwards’ and ‘a lady chef’ but cannot appreciate the interactions between these objects or the overall meaning of the picture. Another simple and useful bedside test for simultanagnosia is using Navon letters (figure 1C)25; patients with simultanagnosia identify local letters but cannot perceive global forms.
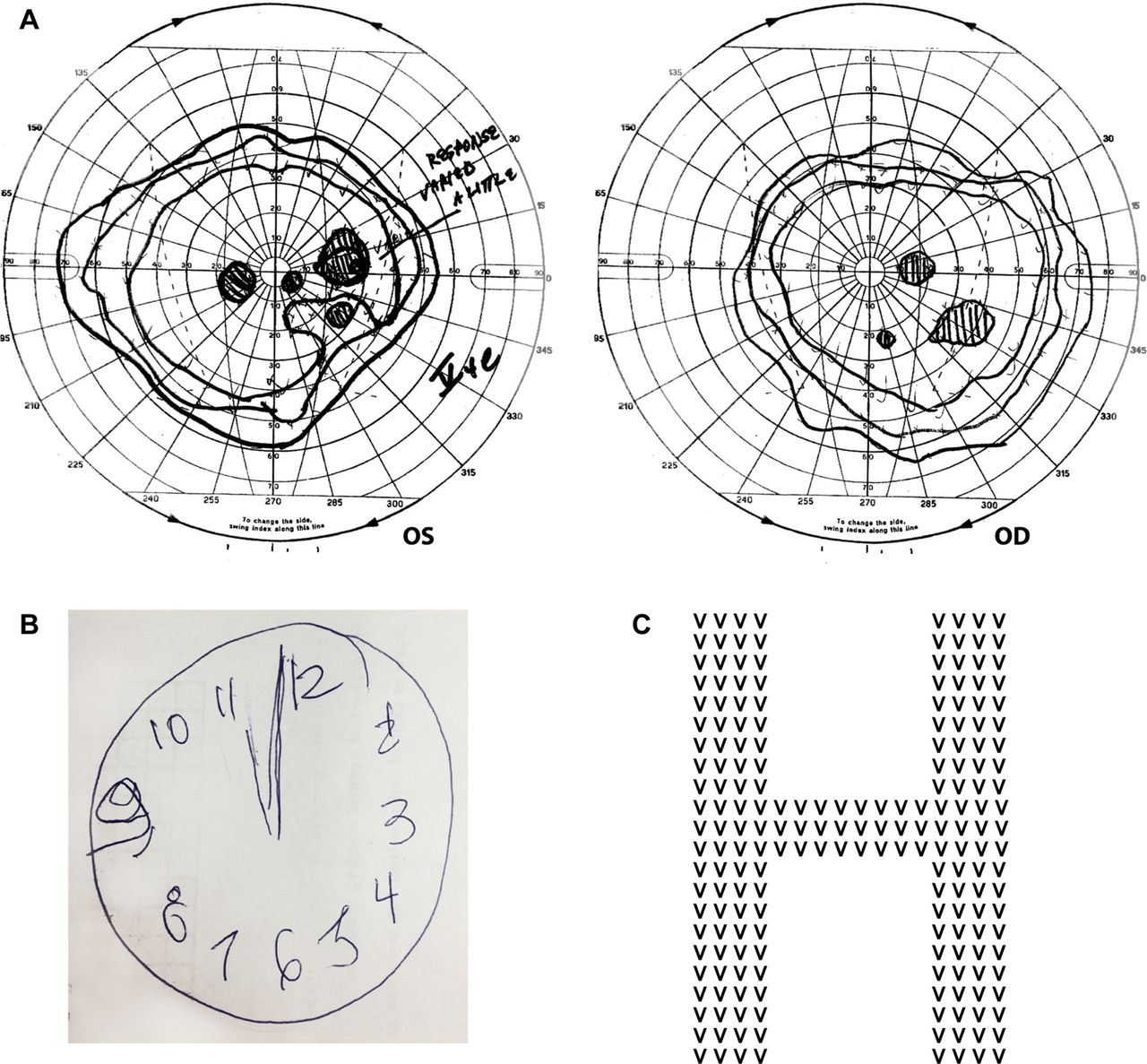
(A) The Goldmann visual field of the patient described in the case vignette clearly shows paracentral visual loss in the right hemifield of both eyes. The inferotemporal field loss in the right eye corresponds to the inferonasal field loss in the left eye and is fairly congruous. However, the inferonasal field loss in the left eye represents a central area of absolute scotoma surrounded by relative scotoma, whereas the corresponding inferotemporal field defect in the right eye represents an absolute scotoma. This suggests incongruity to these otherwise homonymous defects. There is another lesion in the nasal field of her left eye that corresponds to the region of the blind spot in the right eye. The larger-than-expected blind spot in the right eye suggests that there may be a centrocaecal scotoma (a central field suppression that encroaches upon the blind spot). There is also mild, symmetrical, visual field constriction, most prominent temporally, nasally and superiorly. (B) The clock figure drawn by our patient. The clock face is fairly well constructed but she could not set the time to ‘ten past eleven’. (C) A Navon figure: a larger global letter made up of a different, repeating, smaller letter.
In the past, simultanagnosia was considered a failure of fixation disengagement (ie, ‘sticky fixation’). Recent studies, however, have disproved this idea, showing that patients generate more (but smaller amplitude) saccades than controls, making redundant fixations,27 ,32 a finding that we hypothesise to result from impaired inhibition of return (a phenomenon that influences saccadic scan paths whereby novel features take precedence over those recently inspected).33 Furthermore, these patients often fixate on uninformative aspects of a scene and miss salient components that can help them to recognise the global gestalt (figures 2 and 3) (see online supplementary file reference S3). Simultanagnosia is best conceptualised as a restricted window of visual attention.29 Similar to a spotlight, visual attention can be directed to various locations in space, and may be zoomed out to cover a larger area, or zoomed in to focus on a smaller element; just as the spotlight becomes brighter with a narrow beam, attentional acuity is heightened as the window of visual attention is narrowed.29 Dalrymple29 described a patient who eloquently articulated, “My visual field is like a cone that I can extend or shorten. I spend most of my time with a very short visual field concentrating on only one or two things at a time … At times, I have to extend my visual field … This is difficult, because … detail is lost with the extended field, and sometimes everything blends into one”. The ‘expanded spotlight of visual attention’ explains why people with simultanagnosia sometimes perceive the global stimuli but miss local elements. For example, when confronted with Arcimboldo faces (human portraits composed of fruits and vegetables), patients with simultanagnosia can appreciate the global precept of a face but may have difficulty identifying the individual components, possibly because facial stimuli demand an expanded window of visual attention.29 ,30 Although patients with simultanagnosia can perceive a facial form, they may be unable to process the individual's facial features, with inability to recognise them (ie, prosopagnosia). One caveat to remember: attributions of simultanagnosia in PCA must be made cautiously as there may be a restricted effective field of vision without overt simultanagnosia, particularly in older patients, those with right parietal lesions, as well as those with visual field defects (see online supplementary file reference S3).

The MR scan of brain of our case vignette patient showing significant occipital lobe atrophy (especially left sided) with parietal lobe involvement as well. The parietal atrophy contrasts starkly with the relatively well-preserved frontal lobes. In this patient, there were no abnormalities in the basal ganglia or cortical-ribbon on diffusion weighted imaging (not shown here) to suggest the Heidenhain variant of Creutzfeldt-Jakob disease.

{kind=link}
{kind=link}
{kind=link}
An artistic rendering of how patients with simultanagnosia perceive a visual scene. The narrow spotlight of visual attention is directed to local, constituent parts of the scene at the expense of the global gestalt. Additionally, although patients tend to fixate on salient components initially, their attention is subsequently drawn towards uninformative elements of the scene (see online supplementary file reference S3). As a consequence, they fail to integrate these individual elements and create a coherent, visual representation that they can recognise as the iconic Palace of Westminster.
Optic apraxia and optic ataxia
Optic or ocular motor apraxia (which Balint called ‘psychic paralysis of gaze’) refers to the inability to initiate voluntary saccades to visual targets and impaired visual scanning.6 ,33 The main abnormality appears to be impaired visual guidance of saccades, manifest by prolonged latency and inaccuracy, as well as an inability to conduct a visual search of the environment.33 It results from damage to the bilateral posterior parietal cortices, regions important in directing visual attention in the extrapersonal space.33 Optic apraxia can be elicited at the bedside by asking the patient to saccade between two visual targets. Examination will also show impaired smooth pursuit but a normal vestibulo-ocular reflex.
Optic ataxia refers to impairment of goal-directed, visually guided reaching and grasping of targets in the periphery, but with normal movements towards targets in the central visual field, despite normal visual acuity, visual fields, and primary sensory and motor systems.6 ,34 Optic ataxia is due to lesions affecting the region of the intraparietal sulcus.34 At the bedside, the clinician should have the patient fixate on his/her nose while attempting to grasp the clinician's finger when it is presented in the visual periphery. Varying the position of the finger is important to prevent an anticipatory response. Patients with optic ataxia miss and grope for this target; some may reach towards the point of fixation, rather than towards the peripheral target (a phenomenon termed magnetic misreaching).34 The clinician should then have the patient close his/her eyes and attempt to reach for a jingling bunch of keys (ie, acoustically guided reaching), or reach for a memory-guided target; improvement in reaching and grasping abilities with eye closure confirms optic ataxia.
Other visual manifestations
Complex visual hallucinations sometimes occur; their presence—especially if well-formed, non-threatening, or of silent people or animals—suggests that the pathology of dementia with Lewy bodies underlies the PCA.17 ,35 Other visual phenomena of PCA include achromatopsia,8 prolonged colour after images,16 ,36 reverse-size phenomena (accurately perceiving small, but not large, print),16 ,37 the mirror sign (mistaking one's reflected image for that of another person and sometimes attempting to interact with it),35 the illusion of movement of static objects,16 akinetopsia (failure to perceive motion),38 and 180°-upside-down room tilt illusion (possibly an extreme manifestation of visuovestibular mismatch)16 and a peculiar pattern of visual disorientation where patients can track moving targets but cannot localise static objects in 3D space (see online supplementary file reference S3).
Box 2 summarises the visual manifestation features of PCA. Table 1 summarises the proposed criteria for PCA by Mendez et al,7 with subsequent refinements by Tang-Wai et al.8 Box 3 lists the standardised diagnostic criteria following an international collaborative meeting before the 2012 Alzheimer's Association International Conference (see online supplementary file reference S1).
Visual manifestations of posterior cortical atrophy
Glare sensitivity
Simultanagnosia
Optic ataxia
Optic apraxia
Visual field deficits
Complex visual hallucinations
Impaired depth perception
Impaired contrast sensitivity
Alexia
Visual crowding
Prosopagnosia
Palinopsia
Achromatopsia
Prolonged colour after images
Visual disorientation
Reverse-size phenomena
Mirror sign
Illusion of movement of static objects
Akinetopsia
180°-upside-down room tilt illusion
Proposed standardised diagnostic criteria of posterior cortical atrophy (see online supplementary file reference S1)
Cardinal features (all must be present):
-
Insidious onset, progressive course
-
Prominent visual impairments with a relatively intact ophthalmic examination
-
Complex visual dysfunction, for example, components of Balint's/Gerstmann's syndrome, visual field deficits, visual agnosia, environmental disorientation
-
No stroke or tumour
-
Relatively intact insight and memory
Supportive Features:
-
Alexia
-
Onset before age 65 years
-
Ideomotor/dressing apraxia
-
Prosopagnosia
-
Prolonged colour after images
Based on: Crutch et al.41
Proposed diagnostic criteria for posterior cortical atrophy
Work-up
All patients with suspected PCA should undergo neuropsychological testing, and neuroimaging. The verbal IQ and performance IQ components of the Wechsler Adult Intelligence Scale are valuable neuropsychological measures for PCA; patients generally show substantially poorer scores for performance IQ compared with verbal IQ.5 The Hooper Visual Organization Test—where patients must mentally piece together and identify a series of fragmented images—is another useful test for PCA, particularly if there is simultanagnosia.5 Alternatively, the Cortical Vision Screening Test is a useful test of visuoperceptual abilities, and can help detect visual dysfunction in PCA.16
The MR scan of brain typically shows bilateral occipital and parietal lobe atrophy, which may appear asymmetrical.1––3 ,7 ,8 ,11 ,39 There may not always be atrophy in the occipital region, or it may be missed due to anatomical cortical heterogeneity. Furthermore, parietal atrophy is difficult to assess and can be easily overcalled. If the MR scan is apparently normal or ambiguous in suspected PCA, then positron-emission tomography (PET) or single-photon emission CT scans may provide evidence of hypometabolism in this region.3 ,7 ,39 ,40 In PCA due to Alzheimer-related pathology, PET using Pittsburgh compound B may show parieto-occipital amyloid accumulation1; however, this characteristic regional amyloid deposition may only be apparent very early in the course of the disease (see online supplementary file reference S4). Additionally, cerebrospinal fluid analysis can help to determine the cause of PCA by identifying markers of Alzheimer's pathology (total tau, phosphorylated tau on amino acid 181 and amyloid β-42),1 as well as pathologies of, for example, prion disease (protein 14-3-3).
Differential diagnoses
Most cases of PCA are related to Alzheimer's disease-type pathology, with the neuropathological hallmarks of the disease (eg, neurofibrillary tangles and amyloid plaques) principally concentrated in the primary visual cortex and visual association cortex (especially in Brodmann areas 17 and 18, posterior parietal lobe, and cingulate) instead of the memory-related regions.8 ,9 ,11 ,14 Indeed, the International Working Group-2 for Research Criteria for the Diagnosis of Alzheimer's Disease (see online supplementary file reference S2) considers PCA as an atypical form of Alzheimer's disease.
Other pathologies to consider in PCA are dementia with Lewy bodies, corticobasal degeneration and Heidenhain variant of Creutzfeldt-Jakob disease.1 ,8––11 A history of rapid-eye-movement sleep behaviour disorder, complex visual hallucinations, parkinsonism, sensitivity to neuroleptics and diurnal fluctuation of mental status suggest dementia with Lewy bodies. On the other hand, asymmetrical parkinsonism, dystonia, myoclonus, pyramidal signs, and/or sensory dysfunction, as well as motor apraxia, eyelid opening apraxia and alien limb phenomenon favour a diagnosis of corticobasal degeneration.
Heidenhain variant of Creutzfeldt-Jakob disease manifestations may be protean, making it a difficult diagnosis. There may be cortical visual dysfunction, Balint's syndrome and/or Gerstmann's syndrome associated with myoclonus, ataxia, unusual sensations and prominent neuropsychiatric manifestations; it typically pursues a more rapid and relentless course towards dementia and death (usually within a year). Anton's syndrome (cortical blindness)—a feature of the Heidenhain variant of Creutzfeldt-Jakob disease—does not occur in PCA. The MRI often shows the ribbon-like parieto-occipital cortical restricted diffusion characteristic of Creutzfeldt-Jakob disease.
Treatment
While there is no specific treatment for PCA, anticholinesterase medications may help patients with Alzheimer's disease or Lewy body pathology.1 ,13 Early referral to a low-vision centre can potentially help patients learn ways to cope with their visual dysfunction (eg, voice-recognition devices, simplified electronic displays, adjustment of ambient lighting). Physiotherapy and occupational therapy may also help patients develop compensation strategies that can significantly improve their quality of life. The creation of the international PCA Working Party to pool, share and publicise practical, real-world advice and recommendations on treating and managing patients with PCA, as well as promulgate patient and carer support services (see online supplementary file reference S3) is certainly a pivotal step in helping meet the unique needs of this population.
Conclusion
PCA is an uncommon but important clinical syndrome of which neurologists should be aware. Visual complaints often lead patients to seek optometric or ophthalmological evaluation, which is frequently normal, and they are often referred to a neurologist. However, since the visual phenomena of PCA are often non-specific, variable and/or unusual, and because the ocular assessment is normal, and because tests for simultanagnosia are not part of the ‘standard’ neurological examination, and because parieto-occipital atrophy is easily misinterpreted on brain MRI, there is a real danger that these characteristic impairments are dismissed as ‘normal aging’ or even ‘psychogenic’ until their disease is more advanced, delaying appropriate care and advice. Indeed, unless the neurologist is aware of the manifestations of PCA, the physician and patient may be ‘looking but not seeing’.
Key points
-
Posterior cortical atrophy presents with progressive deterioration of visuospatial and visuoperceptual abilities, as well as numeracy and literacy skills.
-
Compared to typical amnestic Alzheimer's disease, posterior cortical atrophy—which most commonly is a variant of Alzheimer's disease—often presents with predominantly visual symptoms at a younger age, with relative sparing of insight, verbal memory and verbal fluency.
-
Simultanagnosia is an important manifestation of posterior cortical atrophy, and should be suspected in patients who cannot read Ishihara's pseudoisochromatic plates but can identify colours. Useful bedside tools to identify simultanagnosia include the Boston cookie-theft picture, Navon figures and Arcimboldo faces.
-
In posterior cortical atrophy that is not due to Alzheimer's disease, clinicians should consider other pathologies, including dementia with Lewy bodies, corticobasal degeneration, and the Heidenhain variant of Creutzfeldt–Jakob disease.
-
All patients with symptoms suggesting posterior cortical atrophy should undergo MR imaging, looking for regional atrophy in the posterior cortices; if the imaging is normal, PET/SPECT may show hypometabolism in this region to support the diagnosis.
Acknowledgments
The authors would like to thank Jason Thean Kit Ooi for preparing the figures and artwork.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online references
Footnotes
-
Contributors SCB was involved in data acquisition, data interpretation, manuscript preparation and manuscript revision. BM and VP were involved in data acquisition, data interpretation and manuscript revision. PC, JH, DZ, and EF were involved in data interpretation and manuscript revision.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Jonathan Schott, London, UK.
Linked Articles
- Editors' commentary
- Editorial
Other content recommended for you
- Here, there and everywhere: higher visual function and the dorsal visual stream
- The language profile of posterior cortical atrophy
- Down syndrome with posterior cortical atrophy
- Qualitative, exploratory pilot study to investigate how people living with posterior cortical atrophy, their carers and clinicians experience tests used to assess vision
- Case of posterior cortical atrophy (PCA) evolved to PCA-CBS
- The topography of metabolic deficits in posterior cortical atrophy (the visual variant of Alzheimer’s disease) with FDG-PET
- Psychoanatomical substrates of Bálint's syndrome
- Homonymous hemianopia as the presenting sign of posterior cortical atrophy
- Clinical assessment of parietal lobe function
- Cerebrospinal fluid biomarkers in the differential diagnosis of Alzheimer's disease from other cortical dementias