Article Text

Statistics from Altmetric.com
Case presentation
An 18-year-old woman was admitted with recurrent seizures. These began focally in the left leg and rapidly generalised. She had complained of a mild generalised headache for the preceding 4 weeks, associated with rainbow-coloured lights in her central vision. On admission, she reported formed visual hallucinations, which included animals and vehicles. She had been previously well, she drank little alcohol, did not smoke, took only an oral contraceptive pill and had no significant family history.
On examination, she was apyrexial. There was continuous myoclonic jerking of the left arm and neck, left homonymous hemianopia, left hemiataxia and left hemisensory loss.
The myoclonus continued despite aggressive antiepileptic treatment with sodium valproate, levetiracetam and clobazam. She was intubated in a thiopentone-induced coma for 24 h.
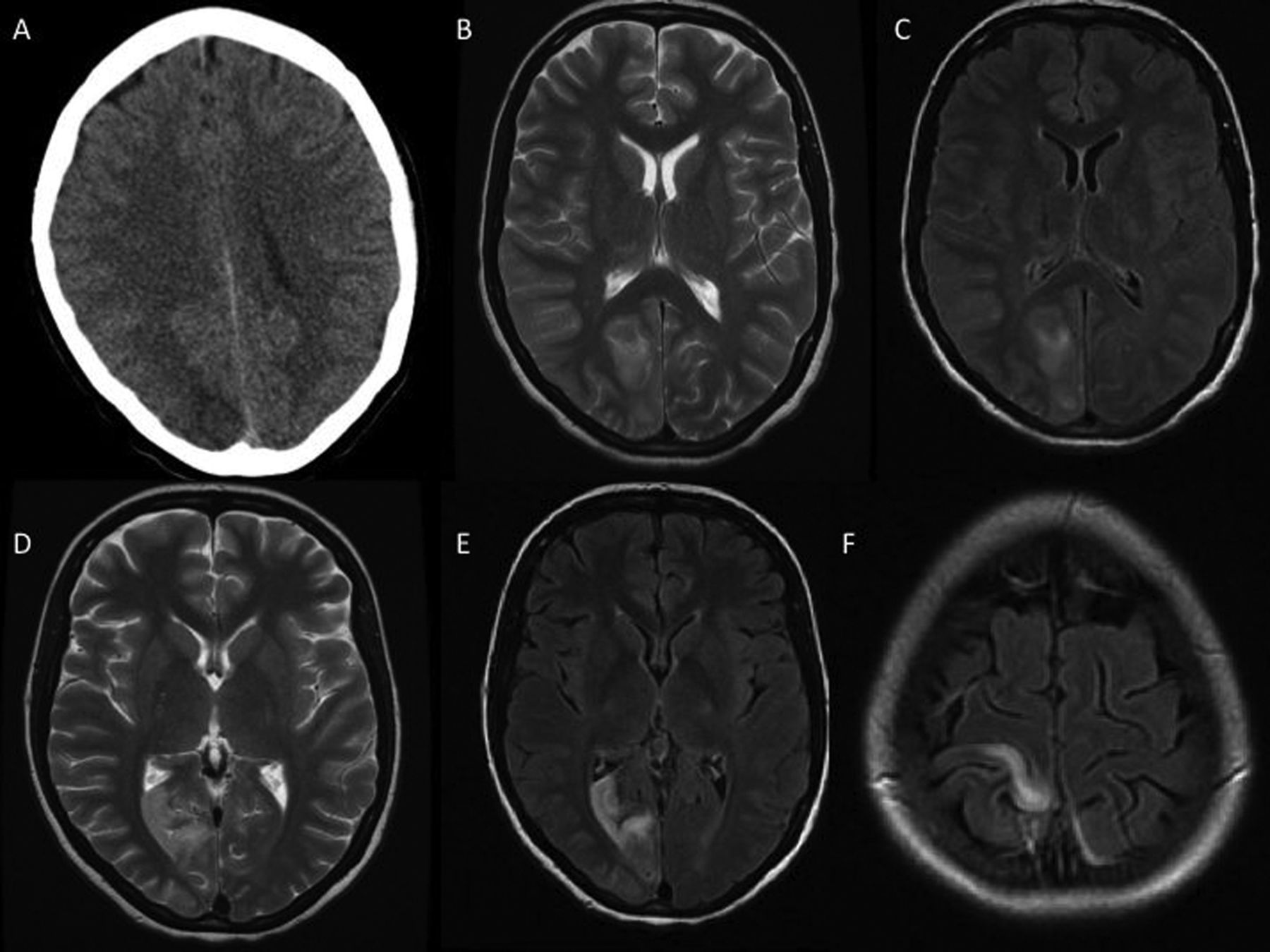
Investigations included normal routine haematology, biochemistry, haematinics and serum lactate dehydrogenase; tests for diabetes mellitus, pregnancy, syphilis and HIV were negative. CT scan of the head showed a right occipital hypodense area, reported as a possible infarct (figure 1A). A CT cerebral venogram was normal. MR scan of the brain showed right occipital and parietal cortical foci of T2 hyperintensity with gyral swelling (figure 1B, C). There was no contrast enhancement or restricted diffusion. Cerebrospinal fluid (CSF) protein was 1.04 g/L (normal ≤0.4) and CSF lactate was 2.5 mmol/L (normal ≤3). The opening pressure, cell count, cytology, glucose, oligoclonal bands, lactate dehydrogenase, viral PCR and cultures were normal or negative. Electroencephalogram (EEG) showed continuous right posterior spike-and-wave activity. MR angiography showed no vertebral artery dissection. Transthoracic echocardiogram was normal. Peripheral neurophysiology showed an axonal sensorimotor neuropathy and myopathic changes.

(A) Axial CT scan of head at presentation, showing a right occipital hypodense lesion. (B) Axial T2-weighted MR scan of brain at presentation, showing a right occipital hyperintense lesion with mass effect. There was also a similar right parietal lesion (not shown). (C) Axial FLAIR MR scan at the same level. (D) Axial T2-weighted and (E and F) FLAIR MR scan 2 weeks later, showing anterior movement of the occipital and medial movement of the parietal lesions.
Two weeks later the myoclonus had continued. Repeat MR scan of the brain showed that the parietal hyperintensity had moved medially towards the longitudinal fissure and the occipital signal was more anterior in the lingual gyrus (figure 1D–F).
We considered an autoimmune encephalitis or malignant infiltration. Her migraine-like headache, progressive myoclonic epilepsy, stroke-like features and predominantly right-sided posterior disease prompted suspicion of mitochondrial disease.
Genetic testing for the mitochondrial DNA (mtDNA) mutations of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) and myoclonic epilepsy with ragged red fibres (MERF) were negative. Nuclear gene testing found that she was homozygous for the c.1399G>A base substitution (p.A467T amino acid substitution) in the polymerase-γ gene (POLG). Both parents were later found to be carriers.
We stopped sodium valproate after a total of 60 days because of the risk of liver failure in POLG disease. Her seizures became well controlled with levetiracetam, eslicarbazepine acetate, perampanel and clobazam. We started ubiquinone and she left hospital after 66 days.
Unfortunately, 30 days later she became confused and jaundiced and the myoclonic jerking restarted. She was apyrexial with a Glasgow coma scale score of 12 out of 15. Inflammatory markers were normal, but liver function had acutely deteriorated, with serum bilirubin 320 µmol/L (1–22), alkaline phosphatase 210 U/L (45–105), alanine aminotransferase 90 U/L (5–35), albumin 19 g/L (37–49), prothrombin time 21 s (11.5–15.5) and activated partial thromboplastin time 49 s (30–40). She was conservatively managed by hepatologists and, because of impaired hepatic metabolism, we stopped eslicarbazepine acetate, perampanel and clobazam, and started topiramate and pregabalin. Over 3 weeks, her liver function improved, she became lucid, the myoclonic jerking resolved and she was once again discharged home. She remains seizure free, liver function is normal and she walks with an aid.
Discussion
Mitochondria are cytoplasmic organelles present in all human cells, with a fundamental role in energy metabolism. They have their own DNA (mtDNA),1 which is maternally inherited. Recent work has shown that the vast majority of the estimated 1500 mitochondrial proteins are encoded by nuclear DNA. This includes those governing replication and maintenance of the mitochondrial genome, such as polymerase-γ, from the POLG gene on chromosome 15q25, which replicates mtDNA.2
POLG mutations are the most common cause of mitochondrial disease currently known, accounting for up to 25% of cases. The carrier frequency of the most common mutation, A467T, is as high as 1% in some European populations.2 The first pathogenic mutation was found in 2001,3 but since then over 150 have been described.4
POLG mutations show a continuous spectrum of clinical features, including epilepsy, myoclonus, ataxia, encephalopathy, ophthalmoplegia, neuropathy and liver dysfunction. However, there are several distinct phenotypes (table 1).4
Clinical phenotypes of POLG mutations
Figure 2 shows a suggested algorithm for genetic testing of POLG mutations. There are no consistent biochemical abnormalities. Serum and CSF lactate and CSF protein may be raised.5 MR scan of the brain may show T2 and FLAIR hyperintense lesions in the occipital lobes (most often on the right), deep cerebellar nuclei, thalamus and basal ganglia, but can be normal, and changes can resolve.6 During active seizures, the EEG often shows an occipital focus. The occipital predilection is not fully understood; the occipital cortex is metabolically active, dealing with high-input visual stimulation, but whether this explains the phenomenon is unproven. Most have an axonal sensorimotor neuronopathy.7

{kind=link}
{kind=link}
Suggested algorithm investigating mitochondrial disease associated with POLG mutations. EEG, electroencephalography; mtDNA, mitochondrial DNA.
Genetic counselling is complex. Though POLG mutations are autosomally inherited, there may be more than one mutation present, genetic factors modify the phenotype, clinical penetrance is unknown for many mutations and carriers may develop late onset mild disease.7 ,8 This case was autosomal recessive and both parents were heterozygous carriers of the A467T mutation. Pregnancy planning can be helped by prenatal diagnosis, either through amniocentesis at 15–16 weeks gestation or chorionic villus sampling at 11 weeks or by preimplantation genetic diagnosis through in vitro fertilisation.
No treatment cures or halts the progression of POLG disease. Most care is supportive and symptomatic. Patients significantly and irreversibly deteriorate after repeated seizures. No single or combination of antiepileptics is superior, but patients often require multiple medications at high doses. Sodium valproate should be avoided at all costs as it can cause fatal liver failure in autosomal recessive disease.
Breakthrough seizures should be treated aggressively. First-line treatments are benzodiazepines such as diazepam or lorazepam, followed by intravenous phenytoin or phenobarbital. If refractory, anaesthesia with propofol or thiopental should be used.6 ,9 Antiepileptic treatment is particularly challenging in liver dysfunction and specialist advice should be sought (table 2).10
Hepatic metabolism of common antiepileptic medications and recommendations in hepatic impairment10
The prognosis of POLG is poor, with most patients dying after prolonged status epilepticus or liver failure. In A467T homozygotes—the most common mutation—epilepsy is the major prognostic factor. In a 10-year study, women were more likely to have epilepsy (75% vs 50% in men) and 59% of people with epilepsy died during the follow-up period.8 A paediatric study found that patients died, on average, 8.5 months after their first seizure.5 People without seizures had a milder course and remained ambulatory over decades.8
Mitochondrial disorders, though rare, will be considered in the differential of a wide range of neurological presentations and in patients with multisystem disorders. POLG mutation is now known to be the most common mitochondrial disease. While there is as yet no cure, greater awareness will improve diagnosis, and neurologists’ involvement in front-line clinical management might influence disease course and prognosis. Aggressive seizure control and the avoidance of sodium valproate are paramount.
Footnotes
-
Contributors MF and KA wrote the article. MF did the literature search. CL supervised the process. CL still looks after the patient.
-
Competing interests CL has received speaker fees and advisory boards from Eisai, UCB and GSK.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed. This paper was reviewed by Patrick Chinnery, Newcastle-upon-Tyne, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Late-onset mitochondrial encephalopathy with lactic acidosis and stroke-like episodes and the role of serial imaging
- Mitochondrial disease: mimics and chameleons
- Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) in the older adult
- Could it be mitochondrial? When and how to investigate
- Frequency of mitochondrial transfer RNA mutations and deletions in 225 patients presenting with respiratory chain deficiencies
- De novo mtDNA point mutations are common and have a low recurrence risk
- MELAS: a new disease associated mitochondrial DNA mutation and evidence for further genetic heterogeneity
- Clinical mitochondrial genetics
- Preimplantation genetic diagnosis for mitochondrial DNA mutations: analysis of one blastomere suffices
- Diagnosis and therapy in neuromuscular disorders: diagnosis and new treatments in mitochondrial diseases