Article Text

Abstract
Every neurologist will be familiar with the patient with atypical spinal cord disease and the challenges of taking the diagnosis forward. This is predominantly because of the limited range of possible clinical and investigation findings making most individual features non-specific. The difficulty in obtaining a tissue diagnosis further contributes and patients are often treated empirically based on local prevalence and potential for reversibility. This article focuses on improving the diagnosis of adult non-traumatic, non-compressive spinal cord disorders. It is structured to start with the clinical presentation in order to be of practical use to the clinician. We aim, by combining the onset phenotype with the subsequent course, along with imaging and laboratory features, to improve the diagnostic process.
- myelopathy
- MRI
- clinical neurology
Statistics from Altmetric.com
Spinal cord dysfunction is a common neurological problem, which may have an obvious traumatic or compressive cause. Beyond this, the lack of pathological specificity of the clinical and imaging features means that patients are often treated empirically, based on the prevalence and the treatability of the differential diagnoses; multiple sclerosis (MS) and isolated inflammatory myelitis are the most likely diagnoses in the western world. Some patients need further investigations if they have atypical features and if these are non-diagnostic the difficulty obtaining a tissue diagnosis may leave the neurologist with a challenging diagnostic dilemma. This article offers a practical approach to the diagnosis of non-traumatic, non-compressive myelopathy in the clinical setting. We focus on disorders that present in adulthood, including metabolic, vascular, inflammatory and autoimmune, neoplastic and infective causes.
Approach to diagnosis
The key features giving most diagnostic value are:
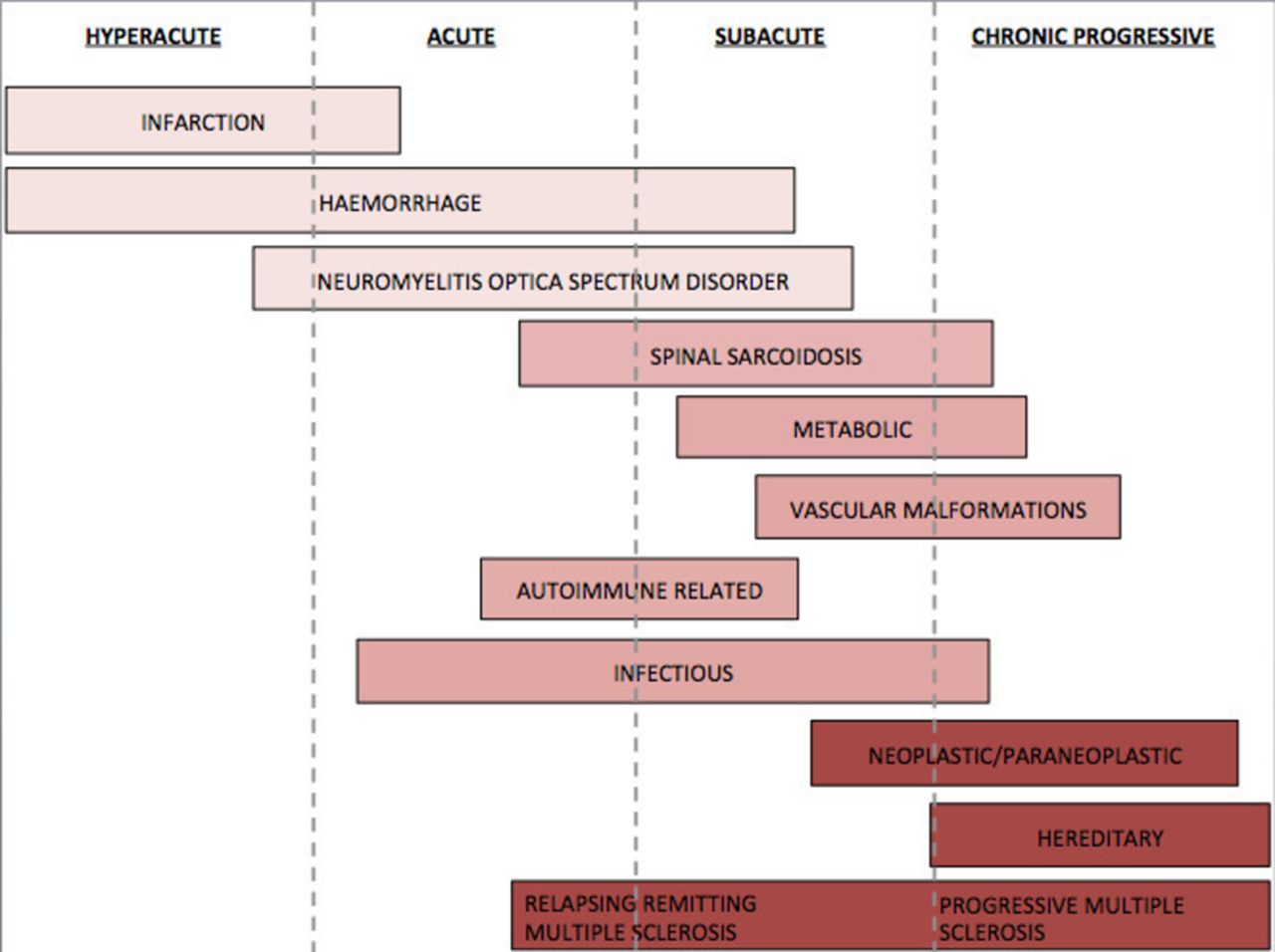
speed of symptom onset (hyperacute, acute, subacute or chronic) (figure 1)
disease course (monophasic or relapsing or progressive, complete partial or no recovery, stable or fluctuating)
lesion appearance on MRI, that is, its length and position, along with the cross-sectional pattern of involvement such as grey or white matter, anterior or posterior or lateral locations, symmetrical or asymmetrical and gadolinium enhancement pattern; the symptoms often reflect these features (table 1)
additional clinical features (table 2).
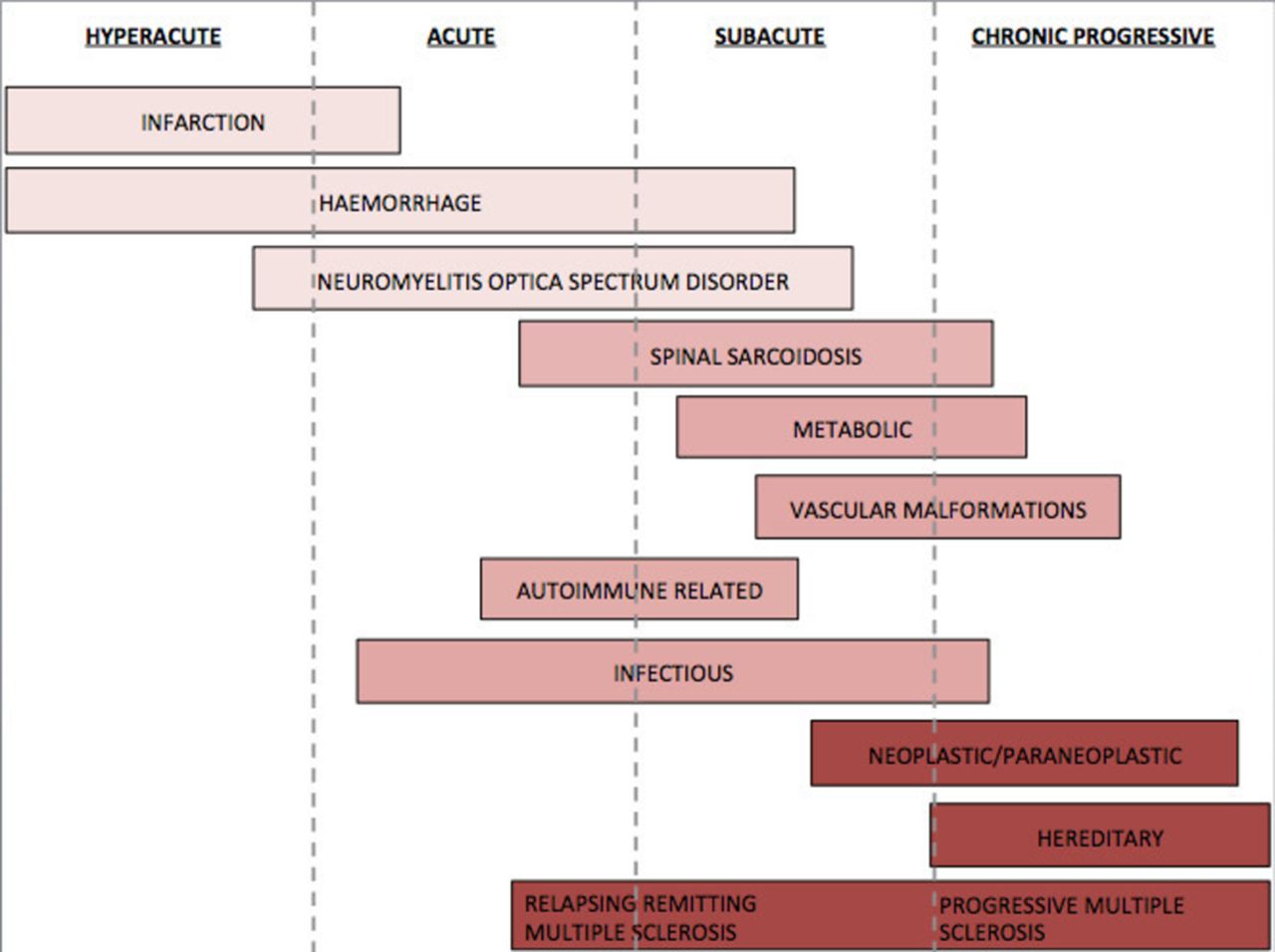
Onset timeline.
Differential diagnosis by speed of onset and lesion length
Characteristics of each differential
Hyperacute onset
Vascular causes of myelopathy (infarction or more rarely haemorrhage) should be suspected when the onset of symptoms is abrupt.
Spinal cord infarction accounts for 1%–2% of all vascular neurological pathology.1 The symptom onset is usually abrupt (within minutes) but the time from onset to nadir may be a few hours. The median time to nadir is around 1 hour but ranges from a few minutes to up to 72 hours.2 The presentation is often associated with flaccid areflexia and mute plantar responses; upper motor neurone signs develop over time. A sensory level is particularly important in this early period to help distinguish this from a peripheral cause. Up to 70% of patients have severe and sudden-onset back pain, typically at the level of the lesion.3 As aortic dissection can cause back pain and secondarily result in lower limb weakness, clinicians might consider CT angiography of the chest to exclude this life-threatening emergency. Two-thirds of patients have an identifiable underlying risk factor,2 4 5 including aortic diseases, aortic surgery, vasculitis, prothrombotic conditions and systemic hypotension.2 4 5 In young people who develop infarction after a Valsalva manoeuvre, consider fibrocartilaginous embolism.
The typical imaging feature, in cases of spinal cord infarction, is T2 hyperintensity in a vascular-specific territory,1 most commonly an anterior ‘pencil-like’ lesion on sagittal sequences and ‘owl/snake-eye’ pattern of signal abnormality on axial sequences corresponding to the anterior horn cells, which are the most vulnerable to ischaemia (figure 2D).5 6 An adjacent vertebral body infarction supports the diagnosis and occurs in 5%–35% of cases.3 5 There is no contrast enhancement in the acute stage but there may be patchy enhancement subacutely.7 Restricted diffusion on diffusion-weighted images appears significantly more sensitive than standard T2 imaging8; however, this feature often does not last for more than 1 week and is more difficult to interpret in the spinal cord than the brain due to technical limitations.9 The initial MRI may be negative and so a normal MRI early on does not exclude spinal cord infarction. In cases of fibrocartilaginous embolism, there may be a disc extrusion adjacent to the site of infarction. Vascular imaging (CT or MR angiogram) is indicated for suspected cervical cord infarction, as vertebral dissection/occlusion is a potential cause.

Typical imaging findings. (A) Spinal cord sarcoidosis with trident sign (A3) dorsal subpial combined with central canal enhancement forming a trident. (B) Spondylosis with enhancement, pancake sign (B2); and typical central grey matter sparing of enhancement (B3). (C) Aquaporin-4 (AQP4)-IgG seropositive neuromyelitis optica spectrum disorder (NMOSD) with short lesion and ring enhancement. (D) Spinal cord infarction showing snake eye (or owl eye) on both T2 and enhancement (rarely seen postgadolinium) in a patient with a spinal cord infarction.
Haemorrhage (intradural or extradural) is a rare cause of hyperacute myelopathy. Spontaneous haemorrhage is uncommon but may occur.10 It is often associated with severe sudden-onset pain that can worsen over hours or up to days after initial onset.11 The diagnosis is based on MRI findings: in the hyperacute phase this consists of a central isointense or hypointense area on T1 imaging with a hyperintense T2 rim. The T1 and T2 signals change with time and provide some information about the age of the haemorrhage.12 T2 signal gradually decreases and T1 signal increases until day 7. Both signals then increase until day 14. Beyond day 14, the signals in both T1 and T2 decrease and the area is ‘dark’. Gradient echo sequences should be used, as spin echo sequences may understate the degree of cord haematoma.13 Intramedullary haemorrhage usually presents as a complication of other conditions that may be acute, subacute, stepwise or chronic.14 The underlying cause, such as spinal vascular malformation (most common) or tumours, may also show on imaging; however, this may be obscured by haemorrhage and further imaging or dedicated vascular imaging may be required to make the diagnosis.15 Other risk factors include a bleeding diathesis or anticoagulation; more rarely there may be a Gowers’ intrasyringal haemorrhage (bleeding into a pre-existing syringomyelic cavity), or haemorrhage as a delayed complication of spinal radiation or a fibrocartilaginous embolism.14
If there is an underlying vascular malformation, both arteriovenous malformations and cavernomas may be the cause. If there is a family history or if there are multiple cavernomas, the patient should be tested for mutations in KRIT1, CCM2 or PDCD10 genes and should have a brain scan.16
Rarely aquaporin-4 (AQP4) positive neuromyelitis optica spectrum disorder (NMOSD) can present hyperacutely and be misdiagnosed as having a vascular cause. Suchdev et al 17 highlighted this when an elderly patient with AQP4 antibodies (AQP4-Ab) presented with sudden-onset transverse myelitis initially thought to be vascular.17
The MRI features of spinal cord infarction and AQP4-Ab positive NMOSD can overlap,18 with no significant differences in lesion length, cross-sectional area and cord expansion. However, 62% of AQP4-Ab positive NMOSD cases have lesions located within 7 cm of the foramen magnum, compared with no cases of spinal cord infarction. Extension to the pial surface, ‘bright spotty lesions’ on axial T2 and gadolinium enhancement are also significantly more common in the AQP4-Ab positive NMOSD group but can occur in up to 30% of cases of spinal cord infarction.18 Additionally, the cerebrospinal fluid (CSF) in people with spinal cord infarction is typically normal.19 However, 53% of patients have a raised protein of up to 0.75 g/L, without pleocytosis or oligoclonal bands.20 In contrast, up to 65% of AQP4-Ab positive NMOSD myelitis cases have a pleocytosis of more than five cells/µl21 and up to 63% have a raised protein (>2.1 g/L) in CSF collected within 30 days of symptom onset.22
Acute/subacute onset
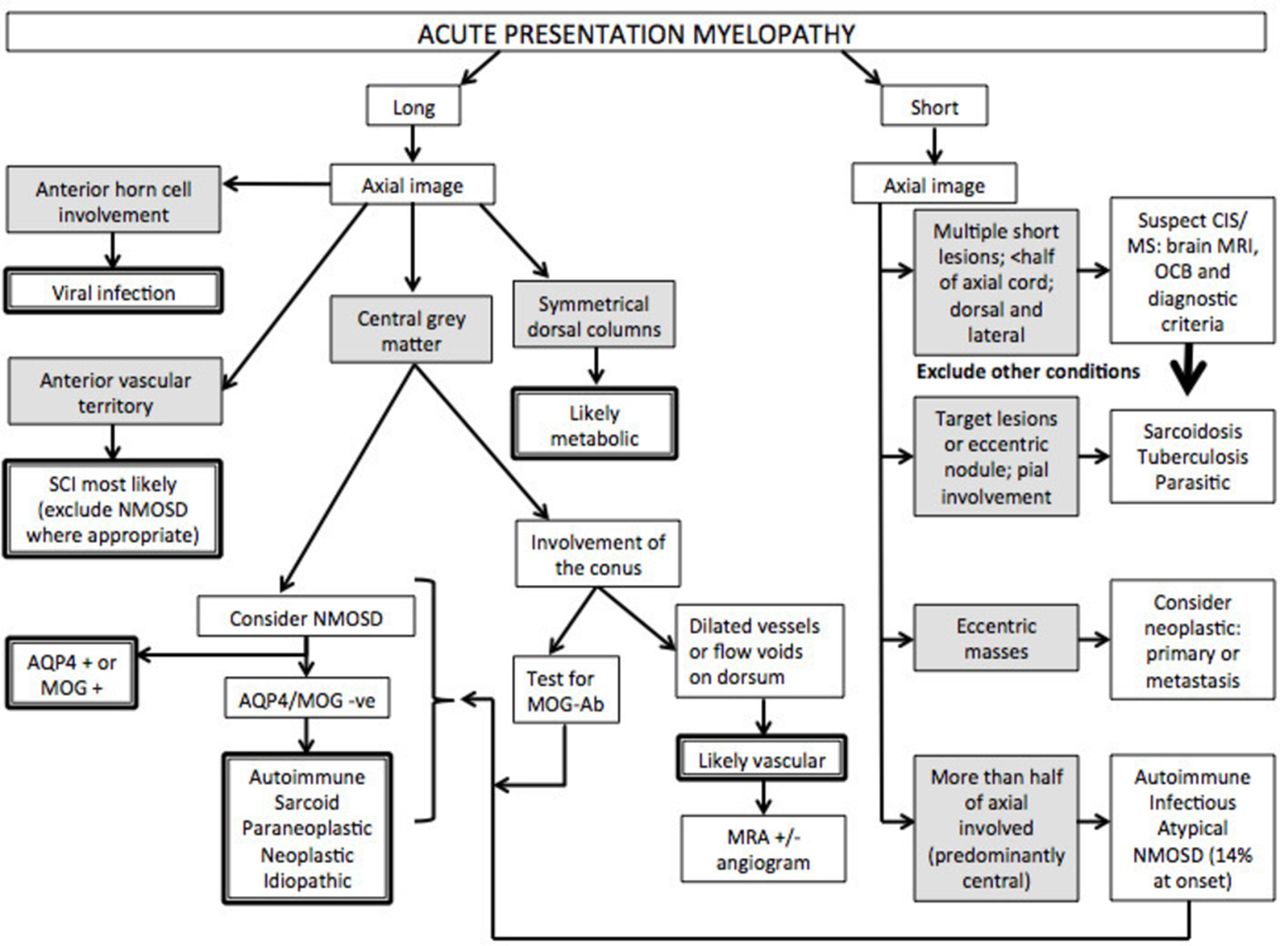
Several spinal cord syndromes have an onset of days to weeks (figure 3). In adults, inflammatory transverse myelitis is the most common. Short spinal cord lesions (<3 vertebral segments) that are partial and asymmetrical on axial views, are classic of MS. Only a few of these cord lesions enhance with gadolinium.23 Patients with these spinal cord lesions who have an abnormal brain MRI have an 83% chance of being diagnosed with clinically definite MS within 10 years, whereas only 11% of those with a normal brain MRI go on to develop clinically definite MS in 10 years.24 The diagnostic criteria for MS also incorporate asymptomatic lesions, highlighting the diagnostic importance of additional asymptomatic spinal cord lesions. An abnormal brain MRI remains the strongest predictor of progression to clinically definite MS (table 3), followed by the presence of oligoclonal bands.25 Oligoclonal bands are positive in >95% of clinically definite MS and in 70%–92% of patients with clinically isolated syndrome.26 27
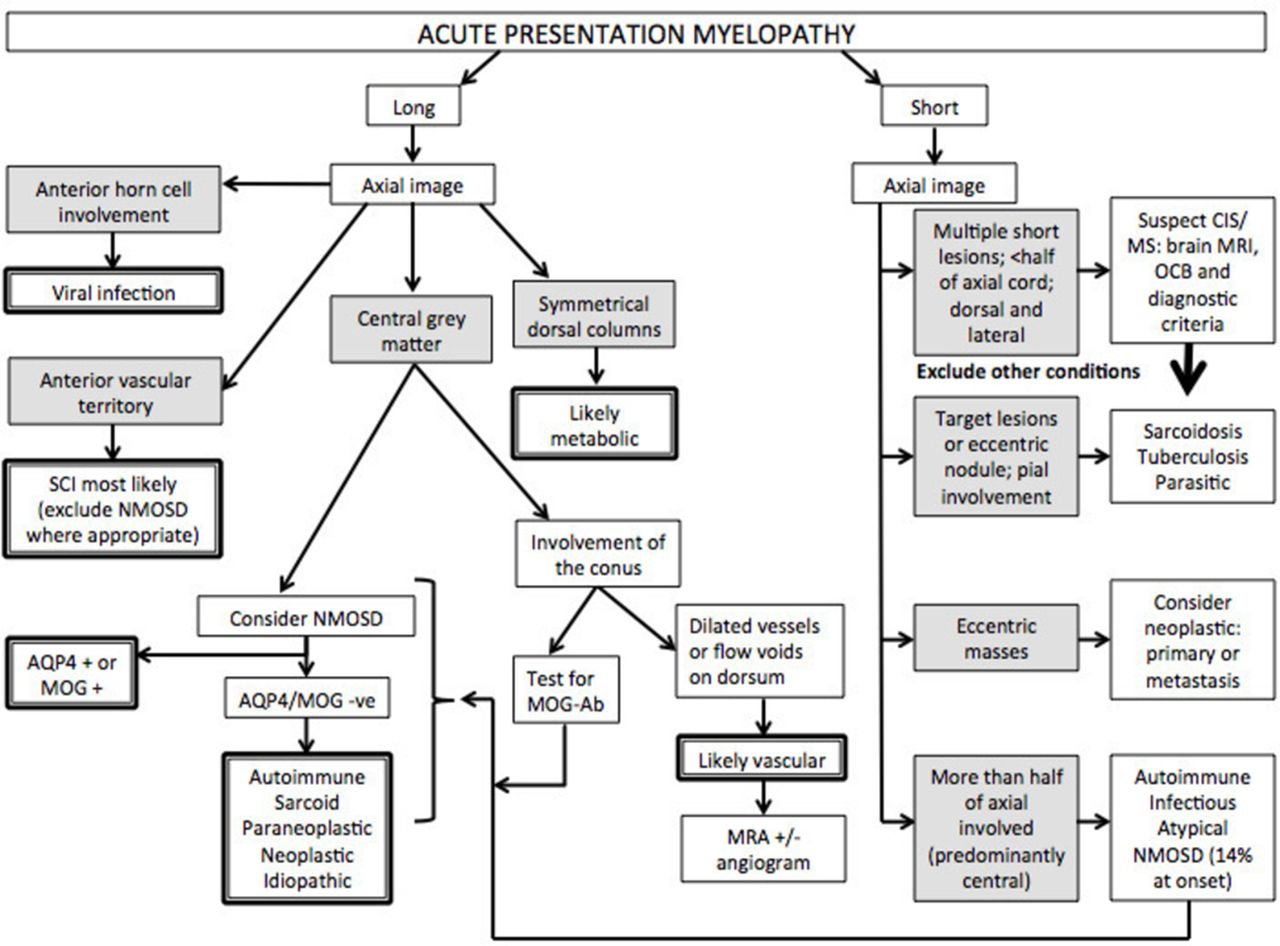
Flow diagram approach to acute/subacute presentation. AQP4, aquaporin-4; MOG, myelin oligodendrocyte glycoprotein; MRA, MR angiogram; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorder; CIS, clinically isolated syndrome; OCB, oligoclonal bands; SCI, spinal cord infarction.
The contribution of brain MRI
Postinfective and infective causes may give longer cord lesions; 30%–60% have an antecedent illness.28 29 This monophasic transverse myelitis is more common in children, with or without concomitant acute disseminated encephalomyelitis. Acute to subacute infective myelitis is most commonly viral and detecting the viral DNA in the CSF may help. It is most important to consider the viruses, such as varicella zoster, for which there is effective antiviral treatment; in contrast, there is no specific treatment for flavivirus and enterovirus-associated myelitis.
Of note, longitudinally extensive transverse myelitis (LETM) of ≥3 vertebral segments can be the first presentation of NMOSD and thus all such patients need to have their serology for AQP4-Ab and myelin oligodendrocyte glycoprotein (MOG) antibody sent; this is preferably done before (or early in) the acute treatment because the serum concentration of antibodies may decrease with immunotherapy and outside of an acute relapse, although they rarely disappear unless low positive. The typical imaging findings include central lesions with grey matter or holocord involvement, usually including the thoracic cord.30 Cervical lesions often extend into the medulla and conus involvement is more frequent in MOG antibody (MOG-Ab) disease.31 However, AQP4-Ab positive NMOSD can present with short lesions (figure 2C) in up to 14% of initial transverse myelitis attacks; the lesions can be eccentric,32 and some are asymptomatic.33 Ring-enhancing lesions (post-contrast) develop in about one-third of patients. This may distinguish AQP4-Ab NMOSD from other causes of transverse myelitis but does not distinguish it from MS.34
A brain MRI may be a useful diagnostic tool in MS but it must be noted that up to 60% of AQP4-Ab positive patients will have white matter lesions; with around 16% (or 27% of those with brain lesions) fulfilling the Barkhof MS criteria for brain lesions35 and so alternative criteria to differentiate MS and NMOSD on brain MRI have been suggested35 (table 3). Although the minority of patients have typical NMOSD brain lesions, they may be highly specific for the diagnosis, affecting the diencephalon and periependymal regions and particularly the area postrema. NMOSD brain lesions outside the common non-specific white matter lesions are usually symptomatic and provide a useful contrast to MS where asymptomatic lesions are characteristic. Of note, area postrema syndromes can be the first presentation of NMOSD and a vomiting illness, subsequently followed by a transverse myelitis, may be misdiagnosed as postinfective. Clues include the length and severity of the vomiting, which may persist for weeks without other gastrointestinal manifestations and may be associated with hiccoughs. Despite CSF oligoclonal bands being much more common in MS than NMOSD (10%–20% in AQP4-Ab NMOSD36 and 13% in MOG-Ab disease37), in the individual patient the presence or absence of oligoclonal bands is of limited diagnostic value.
An important MRI characteristic to consider in the diagnosis of inflammatory myelitis is persistent gadolinium enhancement. Persistent enhancement beyond 3 months should prompt investigation into an alternative diagnosis to MS, NMOSD or autoimmune myelitis.
NMOSD can also mimic spinal cord tumours due to the marked swelling, lesion length, location and intensity, and the diagnosis may only become apparent when biopsied.38 Acute onset and dramatic resolution with corticosteroids and/or plasma exchange supports the diagnosis of antibody-mediated disease. In a recent case of adult-onset biotinidase deficiency mimicking antibody negative NMOSD, the failure to respond to corticosteroids and development of cutaneous lesions prompted a search for a metabolic cause.39
AQP4-Ab NMOSD, more than MOG-Ab disease, may also associate with other autoantibody diseases such as rheumatoid arthritis and systemic lupus erythematosus and may lead to the assumption that these disorders are causative of the transverse myelitis. However, the observation that AQP4-Ab is present in 75% of LETM cases in patients with a definite connective tissue disorder: rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome or systemic sclerosis, which is similar to the percentage expected in NMOSD suggests that the two diseases coexist but are distinct entities.40 41 Therefore, features of NMOSD in any antibody-mediated condition should prompt testing for AQP4-Ab.
Very rarely, Behçet’s disease may be associated with a myelopathy. A recent study has shown that a ‘bagel sign’ on MRI suggests this diagnosis. This sign denotes a central lesion on a T2 axial cut that has a hypointense centre. The hyperintense area also enhances with gadolinium. Sagittal imaging shows lesions of variable lengths. This was noted in 13/14 patients.42
In the appropriate setting, clinicians may consider a metabolic cause, the most common being subacute combined degeneration caused by vitamin B12 deficiency. Of note, the neurological disease may occur either without haematological manifestations43 or as a result of a functional deficiency, which is supported by a raised serum methylmalonic acid or homocysteine and a low concentration of transcobalamin-2.44 The classic MRI finding is a long cord lesion with symmetrical T2 hyperintensity in the posterior and lateral columns, which most commonly involves the thoracic cord. Anterior column T2 hyperintensity and contrast enhancement of the lesion are rare, but can occur in isolated cases.45 46 Clinicians should also suspect this condition in people exposed to nitrous oxide (causing vitamin B12 inactivation), particularly if they had a borderline or low B12 concentration before exposure.47 It is more common following repeated exposure but some people develop subacute combined degeneration after a single exposure.48
Finally, copper deficiency may mimic subacute combined degeneration clinically and on MRI. The differences in imaging of copper deficiency cases, compared with vitamin B12 deficiency, include increased prevalence of cervical cord and central cord involvement in addition to the similar posterior column pathology.49 However, at the individual level these findings are not useful and clinical suspicion is required. Clinicians should particularly consider testing patients who are not responding to vitamin B12 supplements47 or patients with a history of excessive zinc intake.47 A low serum copper and ceruloplasmin would be in keeping with this diagnosis.47 Gastric bypass procedures predispose to both vitamin B12 and copper deficiency. Toxic and metabolic causes, including intrathecal methotrexate, pyridoxine excess and heroin abuse, can also present similarly to subacute combined degeneration.
Inherited metabolic disorders that affect the central nervous system (CNS) can rarely present as a myelopathy in adulthood. The most frequent disorder is the adrenomyeloneuropathy phenotype of X linked adrenoleukodystrophy, which presents in men in their mid-20s (range: second to fifth decades); it occasionally develops in female carriers, but presents later and with a milder phenotype. The typical cord MRI appearances are of thoracic cord atrophy rather than abnormal cord signal.50 Very-long-chain fatty acids are elevated in 99% of affected men, 85% of affected women and 20% of female carriers.51 Genetic testing for mutations on the ABCD1 gene is diagnostic.51 Brain abnormalities occur in around half of men (and less commonly women) and often involve the corticospinal tracts but can be variable and asymmetrical.50
Progressive
There is overlap between the subacute and progressive conditions. Sarcoidosis,52 B12 53 deficiency and chronic infections (eg, human T cell lymphotropic virus myelitis, tuberculosis, schistosomiasis, HIV vacuolar myelopathy and tertiary syphilis) can present with a more slowly progressive picture. A chronic progressive picture excludes NMOSD (figure 4). Progressive MS is the most common cause of a non-compressive myelopathy in the western world, although typically MS leads to a very slowly progressive condition that worsens over decades. However, it is important to note that a compressive myelopathy is sometimes misdiagnosed as inflammatory. It is particularly important to recognise the MRI clues that may assist in the diagnosis of this condition. The ‘pancake sign’ is a transverse-like area of gadolinium enhancement associated with and just below the site of maximal stenosis and at the midpoint of a spindle-shaped T2 hyperintensity (figure 2B). There is often persistent enhancement following decompressive surgery, which may continue for months to years.54
{kind=link}
{kind=link}
{kind=link}
{kind=link}
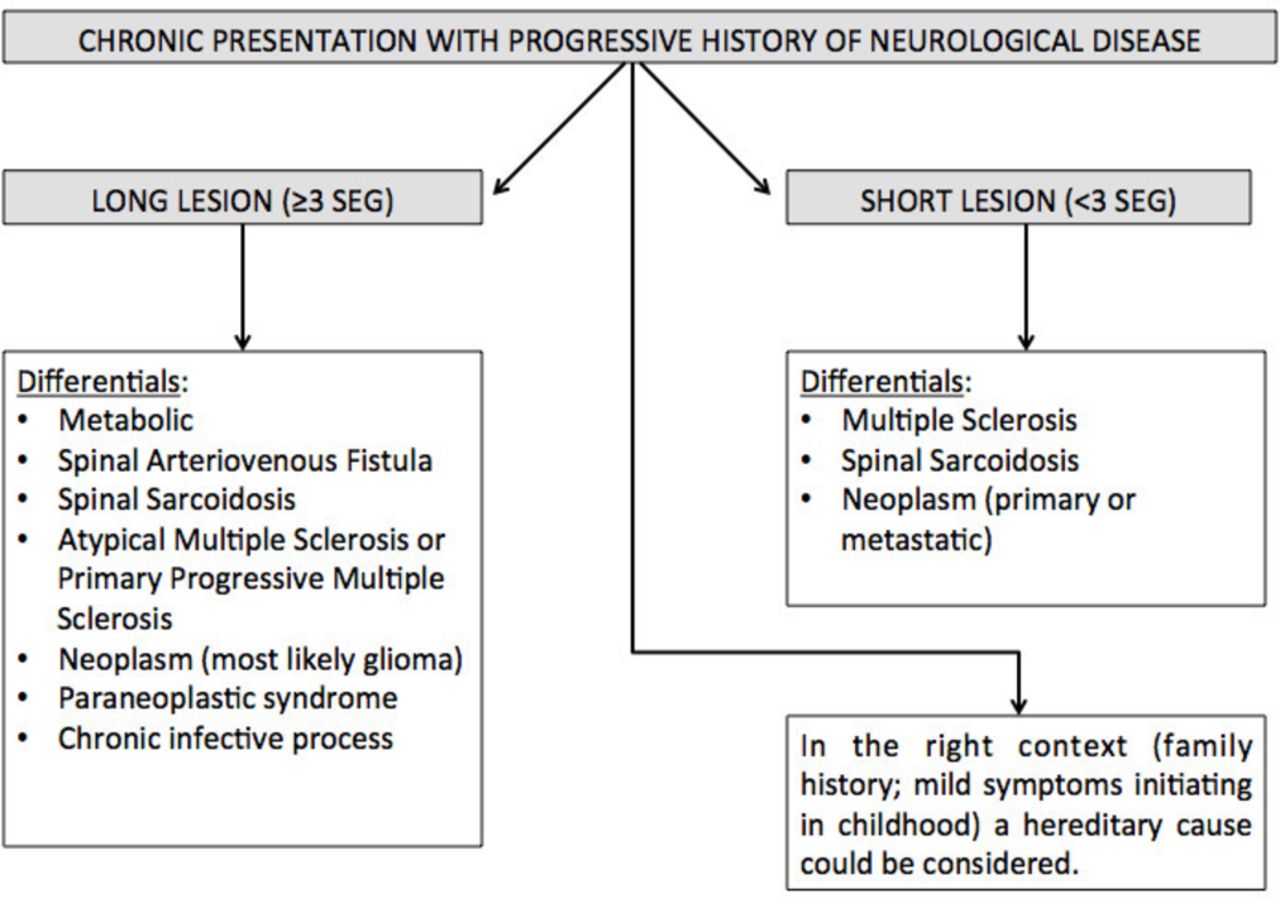
Flow diagram approach to chronic presentation.
Neoplastic processes may very rarely cause myelopathy, affecting the spinal cord by three different mechanisms: primary malignancy, metastatic disease and via paraneoplastic antibody-mediated disorders (discussed separately). Primary intramedullary spinal cord tumours represent 4%–10% of all CNS tumours55 and account for 20% of all intraspinal tumours in adults.56 90%–95% of these are glial tumours, the most common in adults being ependymoma (60%).57 The MRI features of the most common types (ependymoma and astrocytoma) are lesions averaging three to four segments in length (range 1–10), with significant cord enlargement and variable contrast enhancement.58 They occur more commonly in the cervical and thoracic cord but there may be lumbar and conus involvement.58 Up to 4% of patients with a demyelinating disorder or sarcoidosis are initially misdiagnosed as having a malignancy, based on the overlapping MRI features.59 60 AQP4-Ab NMOSD, in particular, can mimic ependymoma; however, in this case report the progression of symptoms in NMOSD was over 6 weeks, which would raise suspicion of an inflammatory rather than neoplastic cause of myelopathy.38 If a neoplasm is suspected, histology is required to diagnose the tumour type.61 62
Metastatic spinal cord malignancy occurs in 0.9%–2.1% of patients with cancer and has a documented mortality of 80% at 3–4 months after diagnosis.63–65 These are distinguished by the MRI appearance of enhanced nodular foci with surrounding oedema, pial enhancement and fludeoxyglucose uptake on positron emission tomography (PET)-CT.63 On MRI, a more intensely enhancing rim around an enhancing lesion (‘rim sign’) and an ill-defined flame-shaped enhancing region at either the upper or lower margin of the lesion (‘flame sign’) help in differentiating metastasis from primary spinal cord malignancies, with a specificity of 94% in isolation and 100% when both are found together.66 They are usually single lesions, but 7% of patients may have multiple lesions. Lung cancer is the most common underlying malignancy (54%), particularly small cell lung cancer, but breast, renal cell, melanoma, lymphoma and, rarely, colorectal cancers may all metastasise to the spinal cord.63 67 Spinal cord metastatic lesions may be the first presentation in up to 39% of patients, but 55% will already have systemic metastasis and 41% will have pre-existing brain metastasis.63 Positive CSF cytology ranges from 18% to 75% depending on the extent of leptomeningeal involvement.68 Other CSF findings are non-specific.
Patients with cancer are also predisposed to postradiation, chemotherapy-related myelitis or infection (often atypical).65 69
Certain disorders are often in the differential but challenging to diagnose
Neurosarcoidosis
Neurosarcoidosis can manifest in many different ways, and encompasses a wide range of pathology including intradural and extradural lesions, intramedullary lesions and arachnoiditis. It may mimic MS, NMOSD, other inflammatory neurological conditions, primary neurological malignancies and metastatic disease.52 70–72 The diagnosis is usually more straightforward in those with known systemic sarcoidosis.
Around 5%–15% of cases of systemic sarcoidosis present with neurological symptoms73 74 and 4%–43% of these have spinal cord involvement52; in those with spinal involvement the myelopathy is often the presenting complaint.52 75 Most of these patients have evidence of systemic disease when investigated, with 60%–90% diagnosed on biopsy of pulmonary or mediastinal lesions,72 74 and other histological specimens from the brain, lymph node or muscle. Isolated involvement of the spinal cord is rare (<0.5% of sarcoidosis cases).76 77 These cases are particularly challenging and all three cases reported in the literature required spinal cord biopsy to make the diagnosis.77–79
MRI findings vary in the spinal cord, but the dominant pattern is linear dorsal subpial enhancement (table 1). Additionally, there may be central canal enhancement, forming a trident on axial images (figure 2A).80 Intramedullary lesions can affect the cervical or thoracic cord73 75 with a mean lesion length of 3.9 segments (range1–9).72 74 Additional supporting features are overlying leptomeningeal involvement72 (especially with meningomyelitis in the context of sarcoid meningitis75), nerve root involvement81 or persistent enhancement of the lesion on follow-up MRI for more than 2 months despite treatment.52 Clues to additional brain involvement are leptomeningeal enhancement, hydrocephalus (if basal meninges involved), cranial neuropathies (particularly VII and VIII) and involvement of the hypothalamic-pituitary axis or, more rarely, the cavernous sinus.75 82
The CSF findings are non-specific.83 Pleocytosis, raised protein and positive oligoclonal bands may all occur, but the CSF may be completely normal even in patients with a positive brain biopsy.84 CSF ACE concentrations lack sensitivity (sensitivity of 24%–55% and specificity of 90%–95%) but if positive may be used to differentiate sarcoidosis from NMOSD.52 However, CSF ACE can also be positive in infective or malignant disease.83 A plain chest X-ray has a higher sensitivity than CSF ACE74 and so chest imaging is essential in suspected cases. A high-resolution CT scan would be the most appropriate chest imaging and may prompt either biopsy and/or bronchoalveolar lavage.85
A whole-body PET-CT scan may help pick up occult disease: it can increase the diagnostic yield86 and is more useful than serum ACE (50% of patients with uptake on PET-CT have a normal serum ACE87). It may also pick up activity in hilar nodes if the CT scan is equivocal. Additionally, PET-CT may find amenable biopsy sites in people with neurosarcoidosis or cardiac sarcoidosis. This includes people with asymptomatic muscle disease who might show increased muscle fludeoxyglucose uptake, allowing subsequent biopsy to confirm the diagnosis.88
Spinal dural arteriovenous fistula
Spinal dural arteriovenous fistulae are easy to miss. Typically, the fistulae are in nerve roots and lead to venous congestion and hypertension of the spinal cord. This results in lesions that ascend rostrally from the conus on serial imaging studies. The fistula itself is commonly located in the thoracolumbar cord, with fewer than 2% of fistulae located in the cervical cord,89 and 4% in the sacral region.90 These account for 70% of spinal vascular malformations.91 The other 30% are due to spinal arteriovenous malformations, which are more likely to present acutely with haemorrhage (see earlier) and commonly occur in the cervical cord.92 In 0.5–4% of patients multiple spinal dural arteriovenous fistulae occur.90
Spinal dural arteriovenous fistulae often have an ascending symmetrical subacute or progressive onset.93 Exacerbations occur with exercise or due to postural change (eg, bending over) and are relieved by rest or changing position.90 94 This may give the impression of a relapsing disease course but the condition continues to progress over time with or without these exacerbations. It is more common over the age of 50 years but may occur in 18 to 91-year-olds, with <1% being under the age of 30.94 95 Due to a variable presentation in the early stages, up to 80% of cases are misdiagnosed on initial presentation,90 91 often as peripheral neuropathies or lumbosacral disease (onset to diagnosis range is 12–44 months).90 The long cord lesions may also mimic NMOSD. Sphincter dysfunction tends to develop over time and once it has developed, it rarely improves after treatment.96 Early diagnosis is important because patients accrue irreversible disability over time. Most patients stabilise after intervention, with only about half of patients reporting improvement in gait and around 30% improvement in sensory symptoms.96 97
Conventional MRI findings show T2 hyperintensity in up to 90% of cases94 with conus involvement in 80%–95% of those.93 94 98 Lesions usually span 3–10 segments93 98 although 15% of patients have single-segment involvement. Dilated perimedullary vessels may show on the dorsum, enhancing with contrast and/or as flow voids on T2 imaging.96 Of note, parenchymal enhancement can lead to suspicion of an inflammatory or neoplastic cause. Therefore, conventional MRI misses half of spinal arteriovenous fistulae and so MR angiography is required (sensitivity of 91% and specificity of 78%).96 Considering that MR angiography may miss 9% of cases, spinal angiography remains the gold standard for diagnosis but an MR angiogram can isolate the levels, and reduce the angiographic contrast load by at least half.96
CSF is usually normal but some cases have a slightly raised protein or mild pleocytosis. In a large review of structural CNS disease over 20 years, there were three patients with a confirmed spinal dural arteriovenous fistula—diagnosed on spinal angiography with recovery after surgical intervention and no evidence of concomitant disease—who had positive oligoclonal bands at first presentation, which delayed their diagnosis.99
Unlike in inflammatory disorders, patients with spinal dural arteriovenous fistulae do not improve with corticosteroids. Furthermore, some patients deteriorate with corticosteroids, which can be an important clue.89 93 100 101
Paraneoplastic
Paraneoplastic neurological syndromes are rare and usually occur in older patients. However, in a study of 31 patients it was noted from as young as 37 years old. They usually present as multifocal disease but some present with isolated myelopathies.102 Paraneoplastic myelopathy may have a subacute or insidious onset102; more rarely, necrotising, rapidly progressive myelopathies may occur.103 104 Up to 81% of patients have an associated antibody, most commonly collapsin response mediator protein 5 and amphiphysin.102 Antineuronal nuclear antibodies 1 and 2, prostate cancer antigen-1 and glutamate decarboxylase-65 also occur in paraneoplastic myelopathy.105 The most common associated cancers are lung and breast, but others include kidney, thyroid, ovarian, lymphoma and melanoma.102 105 Symmetrical lateral tract LETM with variable enhancement is typical but spinal MRI may be normal (one series found this in one-third of patients).102 CSF findings are non-specific: pleocytosis is the most common finding and up to 10% have positive oligoclonal bands.106 This condition can therefore also mimic primary progressive MS; however, the symmetrical findings on MRI and the lack of typical brain features, or a normal brain MRI, should suggest otherwise.105
AQP4-Ab NMOSD may rarely be associated with remote cancers, and should be considered in the elderly.107-109 Older men (>45 years) presenting with LETM were more likely to have an associated malignancy. Thus, this group should be more actively screened for malignancies, in particular for lung and breast, and also prostate, carcinoid tumours and haematological malignancies.108 110 111
The role of biopsy
A tissue biopsy may help in any undiagnosed progressive disease where empirical therapy is not working. For malignant lesions where there are specific treatment implications biopsy (acquired either preoperatively or intraoperatively) is necessary to confirm the diagnosis and its histological type.62 However, due to its eloquent location, spinal cord biopsy is infrequently performed.
There is very little literature to guide the use of spinal cord biopsy in the diagnosis of unknown causes of myelopathy. The Mayo Clinic series of 38 cases of spinal cord biopsy (1988–1998) is the largest cohort studied in the MRI era.112 All patients had presented with progressive neurological disease, isolated to the spinal cord, with non-specific findings on MRI (lesions of variable sizes with patchy contrast enhancement) and CSF (variable levels of pleocytosis and raised protein) and they were considered to have not responded adequately to empirical treatment. Of these cases, 74% subsequently had a biopsy-proven diagnosis, including demyelination, primary malignancy, lymphoma, tuberculosis and schistosomiasis. The biopsies in the remaining 26% showed only non-specific changes. Only 47% of preoperative diagnoses were correct.112
Final thought
There may be cases where a full work-up does not result in a confirmed diagnosis and these patients will usually be treated empirically, often with corticosteroids. It is important to follow up such cases, and to maintain the ‘undiagnosed’ label, in order to keep the mind open to later new diagnostic clues. Neurology in these circumstances can be likened to gardening; watching it grow sometimes reveals its identity.
Key points
The most useful discriminators in diagnosing spinal cord lesions are the local prevalence, the speed of symptom onset and the length of the spinal cord lesion (table 1 and figure 1).
Additional MRI features and, in some cases, specific diagnostic tests, may then narrow down the likely diagnosis (table 2).
Spinal sarcoidosis, spinal arteriovenous fistulae and paraneoplastic syndromes are the most challenging to diagnose; a high degree of clinical suspicion should lead to more extensive testing.
If the diagnosis is uncertain, we recommend treating acutely but avoiding premature diagnostic ‘labelling’; it is best to allow the evolution with time to provide diagnostic clues.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
Footnotes
Contributors RM and JP conceived the review and all authors were involved in the writing of the paper.
Funding RM is undertaking graduate studies funded by The Rhodes Trust.
Competing interests JP: Funding for highly specialised services to run a national congenital myasthenia service and a neuromyelitis service. Support for scientific meetings and honorariums for advisory work from Merck Serono, Biogen Idec, Novartis, Teva, Abide, Chugai Pharma, Alexion, MedDay, Argenx and Bayer Schering, Medimmune and unrestricted grants from Merck Serono, Novartis, Biogen Idec, Chugai, Alexion and Bayer Schering. MS society and Guthie Jackson Foundation research grants. RM is undertaking graduate studies funded by The Rhodes Trust. BGW receives royalties from RSR, Oxford University, Hospices Civil de Lyon, and MVZ Labor PD Dr Volkmann und Kollegen GbR for a patent of NMO-IgG as a diagnostic test for NMO and related disorders. He serves as a member of an adjudication committee for clinical trials in NMO being conducted by MedImmune and Alexion pharmaceutical companies. He is a consultant for Caladrius Biosciences and Brainstorm Therapeutics regarding potential clinical trials for NMO. He serves as a member of a data safety monitoring committee for clinical trials conducted by Novartis.
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Lionel Ginsberg, London, UK.
Other content recommended for you
- Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations
- Myelin oligodendrocyte glycoprotein (MOG) antibody-associated longitudinally extensive transverse myelitis (LETM) and primary Sjogren syndrome: a rare association
- Antibodies to MOG and AQP4 in children with neuromyelitis optica and limited forms of the disease
- Neuromyelitis optica spectrum disorder (NMOSD) presenting as acute transverse myelitis with positive aquaporin 4 antibodies
- Optic neuritis with potential for poor outcome
- Brain structural alterations in MOG antibody diseases: a comparative study with AQP4 seropositive NMOSD and MS
- Early factors associated with later conversion to multiple sclerosis in patients presenting with isolated myelitis
- Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease
- CNS inflammatory demyelinating disorders: MS, NMOSD and MOG antibody associated disease
- Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein