Article Text

Abstract
The physical examination always begins with a thorough inspection and patients with potential neuromuscular weakness are no exception. One question neurologists routinely address during this early part of the assessment is whether or not there is muscle enlargement. This finding may reflect true muscle hypertrophy—myofibres enlarged from repetitive activity, for example, in myotonia congenita or neuromyotonia—or muscles enlarged by the infiltration of fat or other tissue termed pseudohypertrophy or false enlargement. Pseudohypertrophic muscles are frequently paradoxically weak. Recognising such a clinical clue at the bed side can facilitate a diagnosis or at least can narrow down the list of potential suspects. This paper outlines the conditions, both myopathic and neurogenic, that cause muscle enlargement.
- myopathic hypertrophy
- pseudohypertrophy
- neurogenic hypertrophy
Statistics from Altmetric.com
Introduction
Ancient Greek statues were endowed with big muscles, often portrayed in a living contrapposto stance, the measure of all things. Shelley wrote that we are all culturally Greek and therefore perhaps it is unsurprising that modern society contains countless images of subjects who are, in the vernacular, ‘ripped’. Healthy muscles enlarge through exercise but neurologists occasionally encounter patients who have muscle enlargement secondary to neuromuscular diseases. The examination remains a vital part of our routine practice, since slowly changing muscle size may remain unnoticed by the patient and therefore not discussed during the history. Muscle enlargement, the presence of contractures, eliciting positive motor signs such as myotonia, as well as determining the patterns of muscle weakness in our neuromuscular patients are all potential allies in our quest for a bedside diagnosis and can help to shape subsequent investigations. Clinical examination is also particularly important when assessing weakness, since motor signs frequently precede motor symptoms, whereas sensory symptoms usually precede sensory signs.
If we do discover muscle enlargement, what neurological conditions should we suspect and how should we investigate it? This paper aims to provide some answers.
I will briefly describe some aspects of the physiology of normal muscle hypertrophy before describing the neurogenic and myopathic conditions that can lead to both hypertrophy and pseudohypertrophy.
True skeletal muscle hypertrophy
Muscle growth is regulated by signal transduction pathways. These pathways sense and compute local and systemic signals and regulate various cellular functions. Skeletal muscle needs to be able to ‘decide’ to hypertrophy by ‘sensing’ a new exercise demand, integrating this need with other potentially conflicting pieces of information, including how much energy is available for growth. Muscles are aware of mechanical stimulation (mechanotransduction) through conformational changes to proteins during muscle contraction and relaxation and can quantify energy reserves, using, for example, the availability of AMP as a surrogate marker with AMP-activated protein kinase.1
Hypertrophy will therefore precisely match any physical challenge the body encounters, including the asymmetric humeral hypertrophy evident in Neanderthal remains that probably reflected long hours of hide scraping—a strenuous Neanderthal version of ‘washing-up’—rather than any romantic notion of spear use.2
Neurogenic muscle enlargement
It seems heretical to suggest that neurogenic disorders (diseases of motor neurones or their axons) might cause muscle enlargement; we instinctively know that muscle atrophy accompanies neurogenic disorders and have done so since our first faltering steps in neurology as medical students.
However, we undoubtedly do occasionally see muscle enlargement, both hypertrophy and pseudohypertrophy, in neurogenic disorders; so what could be the possible explanation?
In those neurogenic disorders characterised by continuous muscle fibre stimulation—whether myokymia, neuromyotonia or complex repetitive discharges (complex repetitive discharges, pseudomyotonia)—it is clearly an appealing explanation that myofibre hypertrophy might follow, since such continuous muscle activity represents a form of ‘internal physiotherapy’. Complex repetitive discharges can develop in radiculopathies, explaining some, though not all, cases of calf hypertrophy secondary to an L5 or S1 radiculopathy. If the ‘internal physiotherapy’ is curtailed, with carbamazepine or intramuscular botulinum toxin, then not only is the positive motor activity reduced or stopped but the accompanying muscle hypertrophy also reduces, further supporting the hypothesis.3–5
However, neurogenic hypertrophy also develops without continuous motor activity. In these neurogenic disorders, intact type II myofibres in partially denervated muscle probably hypertrophy in response to their increased workload; in addition, stretch can cause type I myofibre hypertrophy even when denervated.6 7 This myofibre hypertrophy therefore accounts for the overall increase in size of the denervated muscle; however, such hypertrophy fails to compensate fully for the presence of completely denervated atrophic myofibres within the same muscle, and so such enlarged muscles are paradoxically weak.6
There are also times when neurogenic muscle enlargement reflects infiltration by fat or collagen (pseudohypertrophy).5
It might help to draw a distinction between asymmetric focal neurogenic hypertrophy and more symmetrical neurogenic disorders, as they have different causes.
Focal asymmetric neurogenic hypertrophy
Patients with focal, asymmetric muscle enlargement complicating radiculopathies (or more rarely old poliomyelitis) might present to a general neurology clinic. In some cases, there is true hypertrophy as outlined above but in other cases, particularly old poliomyelitis, the muscle enlargement can reflect pseudohypertrophy.5
Table 1 summarises the causes of asymmetric focal muscle enlargement which include both neurological and non-neurological conditions.
Focal causes of asymmetric muscle enlargement
Focal myositis is a rare and confusing disorder. It is characterised by focal muscle swelling, minor elevation of creatine kinase and inflammation as well as denervation changes that are visible pathologically. It is often linked to a radicular, peripheral nerve or plexus irritation, although importantly it might occur without pain. It might therefore represent a neurogenic disorder, where denervation initially causes some myofibre hypertrophy (see above) and then later myofibre splitting followed by necrosis. The necrosis elicits an inflammatory response including macrophage invasion, and hence the term focal myositis.8 9 Giant cell myositis (granulomatous myositis) is mentioned here for completeness; it is not a neurogenic disorder but a form of inflammatory myopathy that can cause foal muscle enlargement.10
More symmetrical neurogenic hypertrophy
More symmetrical muscle enlargement, often of the calves, can rarely develop in Charcot-Marie-Tooth disease. However, most neurologists are more likely to encounter it in someone with spinal muscular atrophy.
Spinal muscular atrophy is a term understood to imply isolated loss of spinal motor neurones with consequent neurogenic weakness and usually muscle atrophy. The most common that adult neurologists encounter is type III autosomal recessive spinal muscular atrophy. This condition subdivides into different types according to age of onset and natural history: people with spinal muscular atrophy III can walk initially and have a normal lifespan (type IIIa has onset before age 3 years; type IIIb after age 3 years).
Autosomal recessive spinal muscular atrophy is caused by a disruption in the survival motor neurone gene (SMN1) on chromosome 5, most commonly a homozygous deletion of exon 7.11 The SMN1 protein is essential for life with a critical role in assembly of small nuclear ribonucleoproteins (snRNPs), which are involved in pre-mRNA splicing. Figure 1 outlines the process of pre-mRNA splicing, which is also important in understanding the dystrophinopathies outlined later.

Genetic splicing and the dystrophin ‘reading frame’.33
The part of chromosome 5 that contains the SMN1 gene underwent a duplication event approximately 6 million years ago. Thus, chromosome 5 contains a copy of the SMN1 gene—termed SMN2—which is almost identical to SMN1 although differing by a few nucleotides.12 One nucleotide change in exon 7 disrupts a modulator of splicing and means that approximately 80%–90% of mRNA transcript from SMN2 does not include exon 7. The resulting truncated protein is rapidly degraded. The small amount of SMN protein produced cannot fully compensate for the loss of SMN1 gene.
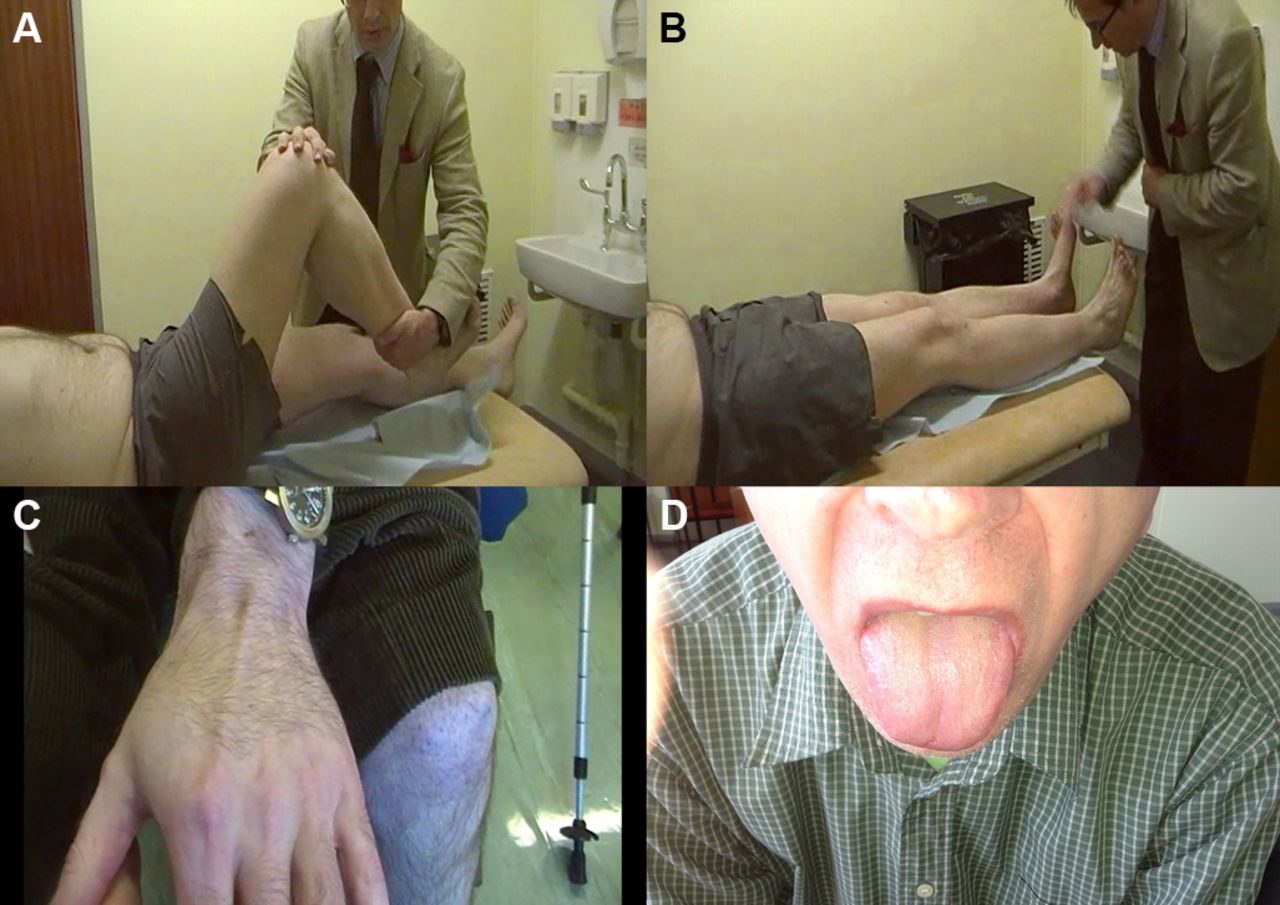
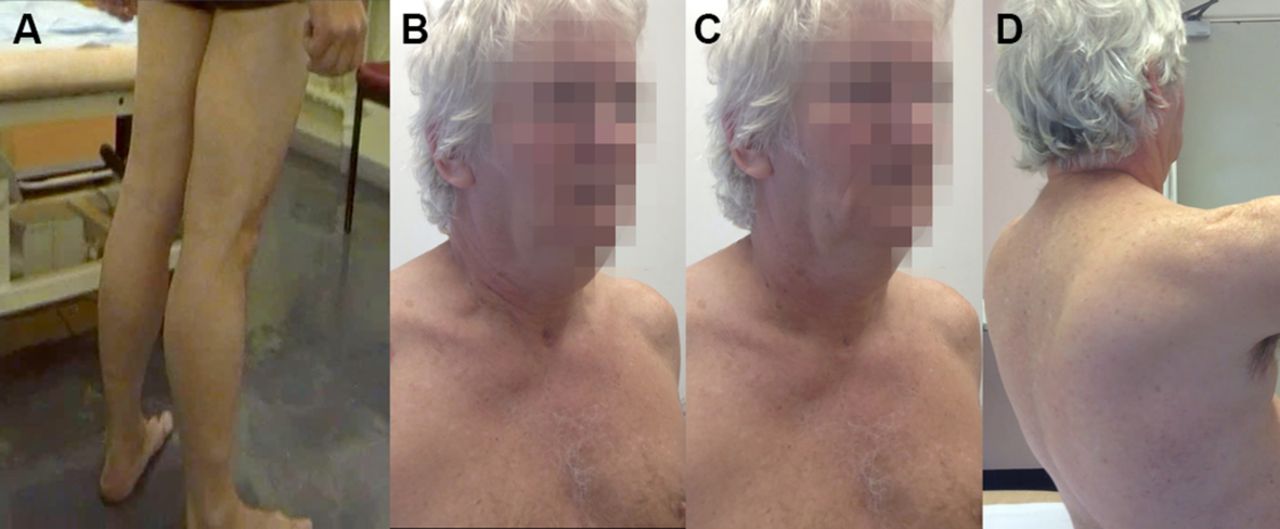
Autosomal recessive spinal muscular atrophy is characterised clinically by a proximal weakness, raised serum creatine kinase and muscle hypertrophy. These clinical features overlap with several muscle diseases, notably Becker’s muscular dystrophy (described below). Clearly, cases of mistaken identity may follow.13 Figure 2 describes other clinical clues that allow us to distinguish between them.

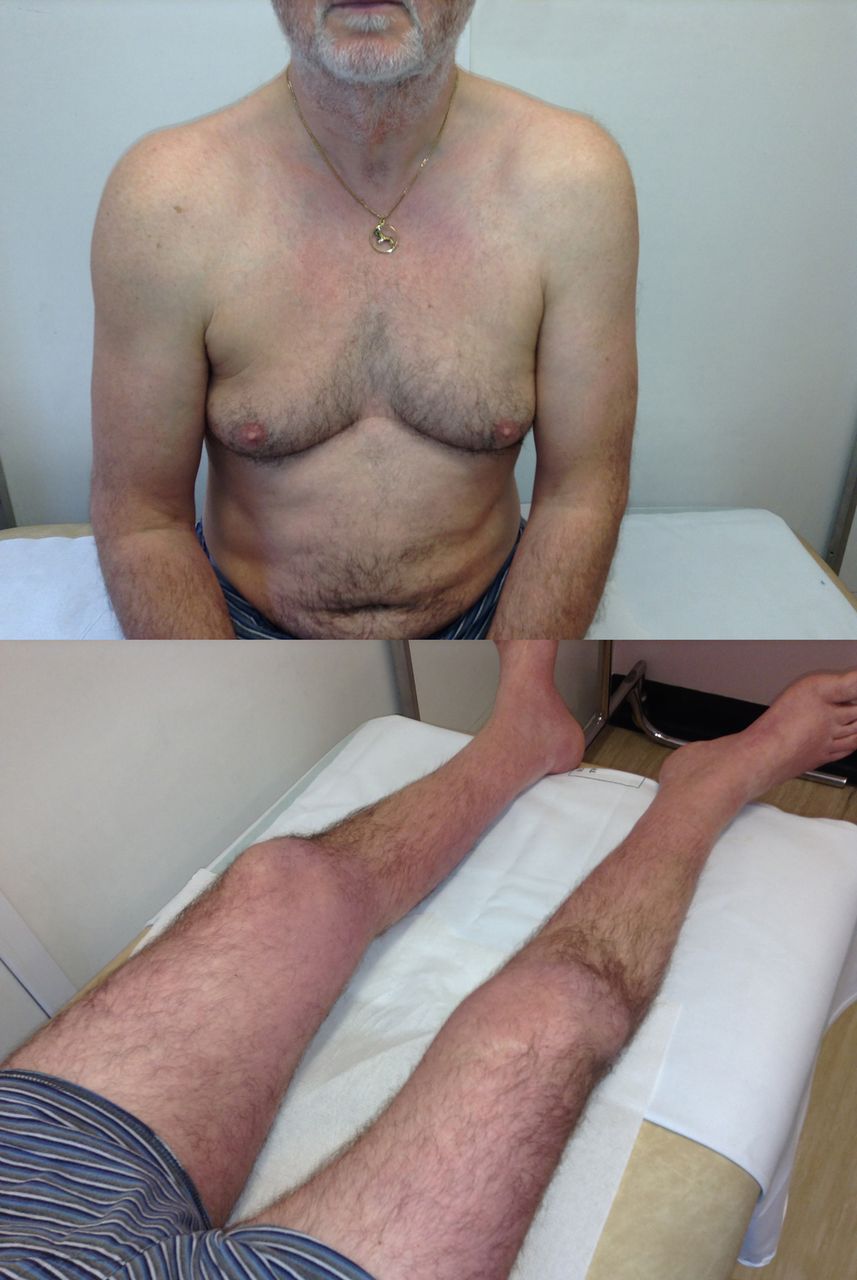
The person in (A) has spinal muscular atrophy type III and the person in (B) has Becker’s muscular dystrophy. Both have enlarged calves and both have hip flexion weakness; these conditions have a similar age of onset and a similar range of raised serum creatine kinase levels. Distinguishing these conditions starts clinically. In spinal muscular atrophy, there are typically fasciculations. In Becker’s muscular dystrophy, there is weakness in biceps as well as triceps and weakness of hip extensors as well as hip flexors, whereas in spinal muscular atrophy, the weakness is more marked in triceps and hip flexors with relatively preserved biceps and hip extensors. Electromyography can help to distinguish a neurogenic disorder from a myopathic one (C and D). There are many causes of calf asymmetry. Take a good look. In this case, a man who had recently started cycling (friends had then noted his calf asymmetry) clearly had a very long-standing condition, since he also had a smaller right shoe size and on reflection he felt that there had always been some asymmetry in calf and leg size.
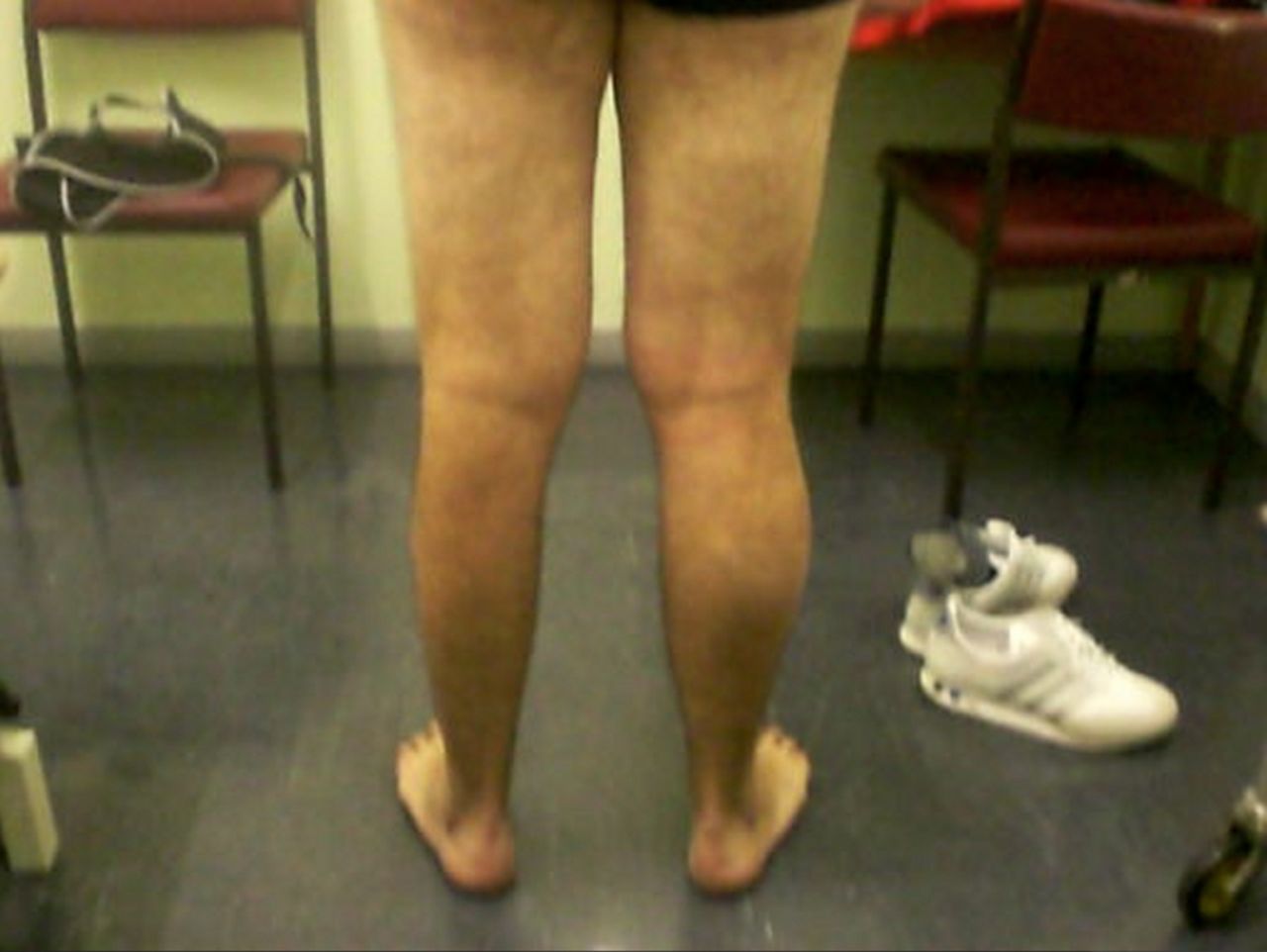
There are several different genetic conditions that fall within the spinal muscular atrophies umbrella, which neurologists will occasionally encounter in clinics. Spinal muscular atrophy IV, with onset after the age of 30 years, is genetically heterogeneous (approximately 50% SMN mutation). Muscle hypertrophy or pseudohypertrophy also develops in some of the other genetic forms of spinal muscular atrophy, for example, spinal muscular atrophy-lower extremity dominant (SMA-LED) (see figure 3).
Another form of spinal muscular atrophy caused by a mutation in the dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) that can cause spinal muscular atrophy-lower extremity dominant (SMA-LED). The clinical spectrum of DYNC1H1 is wide and can also cause a form of axonal Charcot-Marie-Tooth disease and sometimes a central nervous system neuronal migration defect.34 This man had weak wasted legs from the very start of his life but remained ambulant. It is very striking that his main weakness in his quadriceps meant that he could barely overcome gravity—often a clinical clue to SMA-LED. In contrast, his upper limb muscles were bulky, although with occasional fasciculations. MR imaging of the lower limbs often find relative hypertrophy of the adductor longus and semitendinosus; muscle histopathology does not invariably show classic neurogenic features. An electromyogram found chronic neurogenic changes with normal sensory action potentials. Dynein is a motor protein that, among many other functions, ‘walks’ along the microtubules transporting cellular cargo from the cell periphery to its centre.
Muscle disease and muscle enlargement
Muscle hypertrophy more commonly occurs in the setting of muscle disease.
Dystrophinopathy
The dystrophinopathies are X-linked recessive conditions that include the best known forms of muscular dystrophy: Duchenne’s muscular dystrophy (incidence 1:3600 live male births) and its milder allelic cousin Becker’s muscular dystrophy (one-third the incidence of Duchenne’s muscular dystrophy).
Dystrophin is a large 427 kDa protein localised to the sarcolemma and is an important component in a protein complex (dystrophin-associated protein complex) that links the intracellular cytoskeleton to the extracellular matrix. This ‘scaffolding’ complex appears to play a structural role in ensuring membrane stability and force transduction during muscle contraction. The most common form of mutation is a deletion within the dystrophin gene on Xp21 (65%–70% of Duchenne’s muscular dystrophy, 80% of Becker’s muscular dystrophy) although duplications and point mutations also occur.14
The major determinant of disease severity is not the size of deletion in the Duchenne’s muscular dystrophy gene but whether the reading frame is retained. If the reading frame is maintained then a shortened (but functioning) dystrophin protein is translated, as opposed to a disrupted reading frame causing a premature termination and often complete loss of dystrophin (see figure 1).14 15 The serum creatine kinase in Duchenne is usually 40–60 times the upper limit of normal (>10 000 IU/L; normal 55–170 IU/L) and in Becker 5–20 times above normal.
Patients with Becker’s muscular dystrophy retain significant amounts of partially functional dystrophin protein on biopsy. Their first symptoms often appear at age 6–18 years (50% by age 10 years) with mean age of ambulatory loss in the fourth decade. We are likely to see patients with Becker muscular dystrophy in our clinics. A progressive dilated cardiomyopathy commonly evolves, bearing no correlation to skeletal muscle weakness, and so cardiac surveillance in those patients with normal hearts is generally repeated every 3 years.
Muscle enlargement in dystrophinopathies has traditionally been attributed to deposition of fat and connective tissue, hence the term pseudohypertrophy. Imaging techniques, however, have led to a growing appreciation that as well as pseudohypertrophy there might be a true increase in muscle mass. Myofibre hyperplasia (increase in cell number) and myofibre hypertrophy may both occur histologically throughout the natural history of Duchenne’s muscular dystrophy.16–19 Other muscles besides the calves do occasionally enlarge, including, for example, sartorius, tongue (macroglossia) or hand muscles.
Limb-girdle muscular dystrophies
The differential diagnosis of Becker’s muscular dystrophy in the adult neurology clinic includes the limb-girdle muscular dystrophies (LGMD).
LGMD2I is one of the three more commonly encountered in UK practice and is frequently characterised by calf pseudohypertrophy. The other two common LGMD are LGMD2A (calpainopathy) and LGMD2L (anoctaminopathy). LGMD2A more often causes atrophy and the presence of pseudohypertrophy is a useful clinical clue pointing away from a diagnosis of LGMD2A, even when other investigations raise it as a possibility.14 20
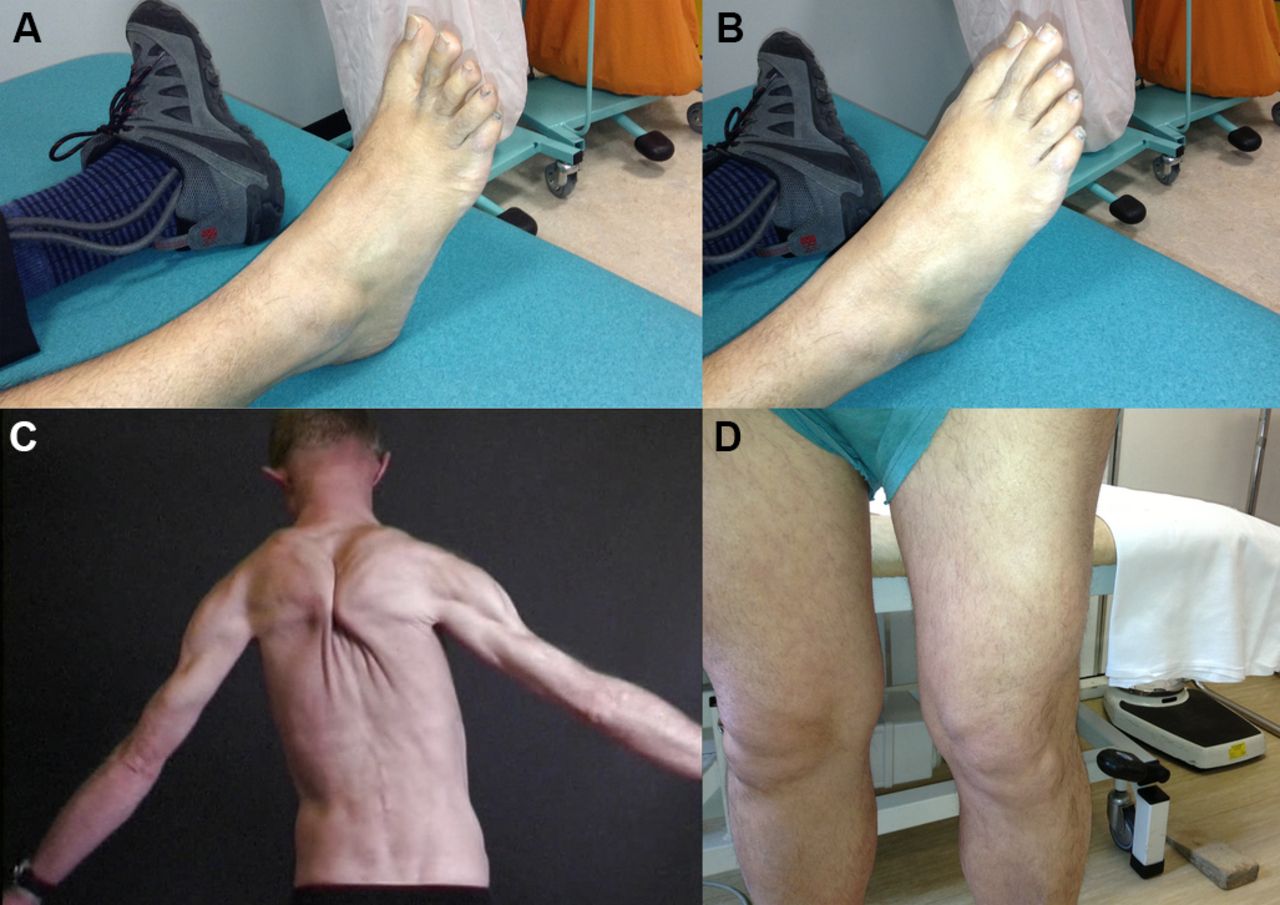
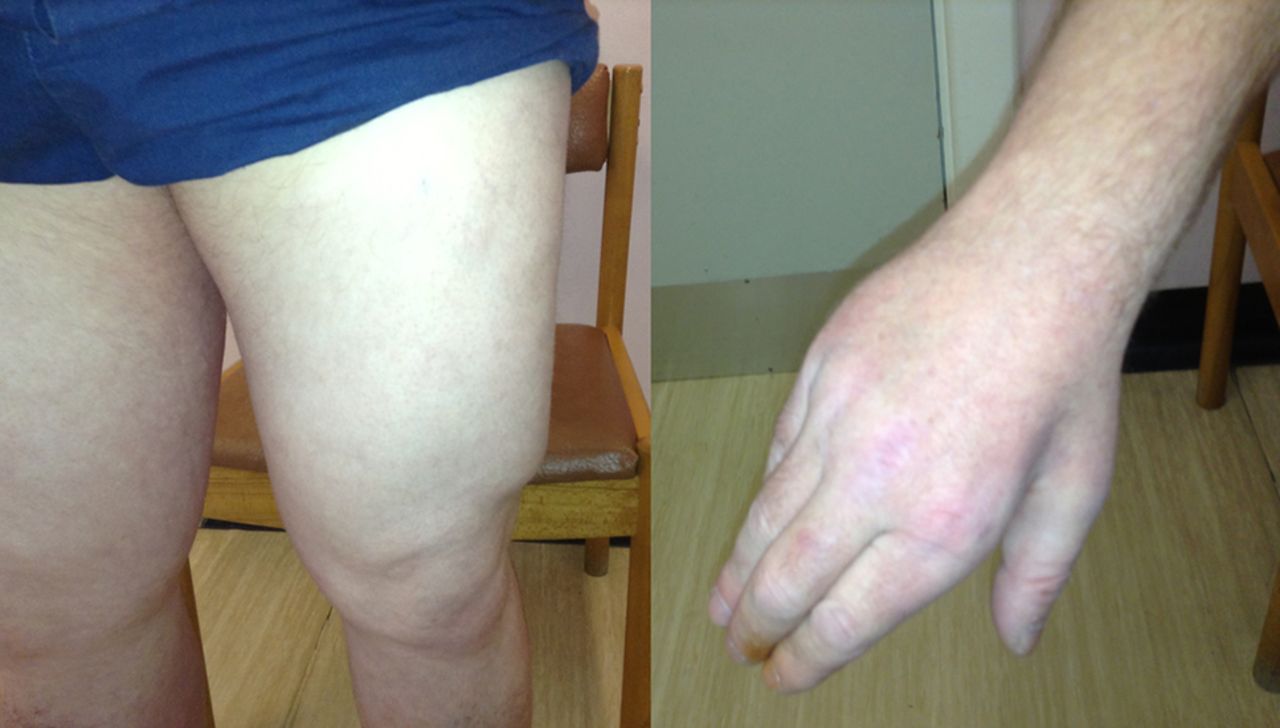
LGMD2L can cause calf hypertrophy particularly early on in its natural history but more frequently it is characterised by muscle atrophy, often in a strikingly asymmetric pattern (see figures 4–6).
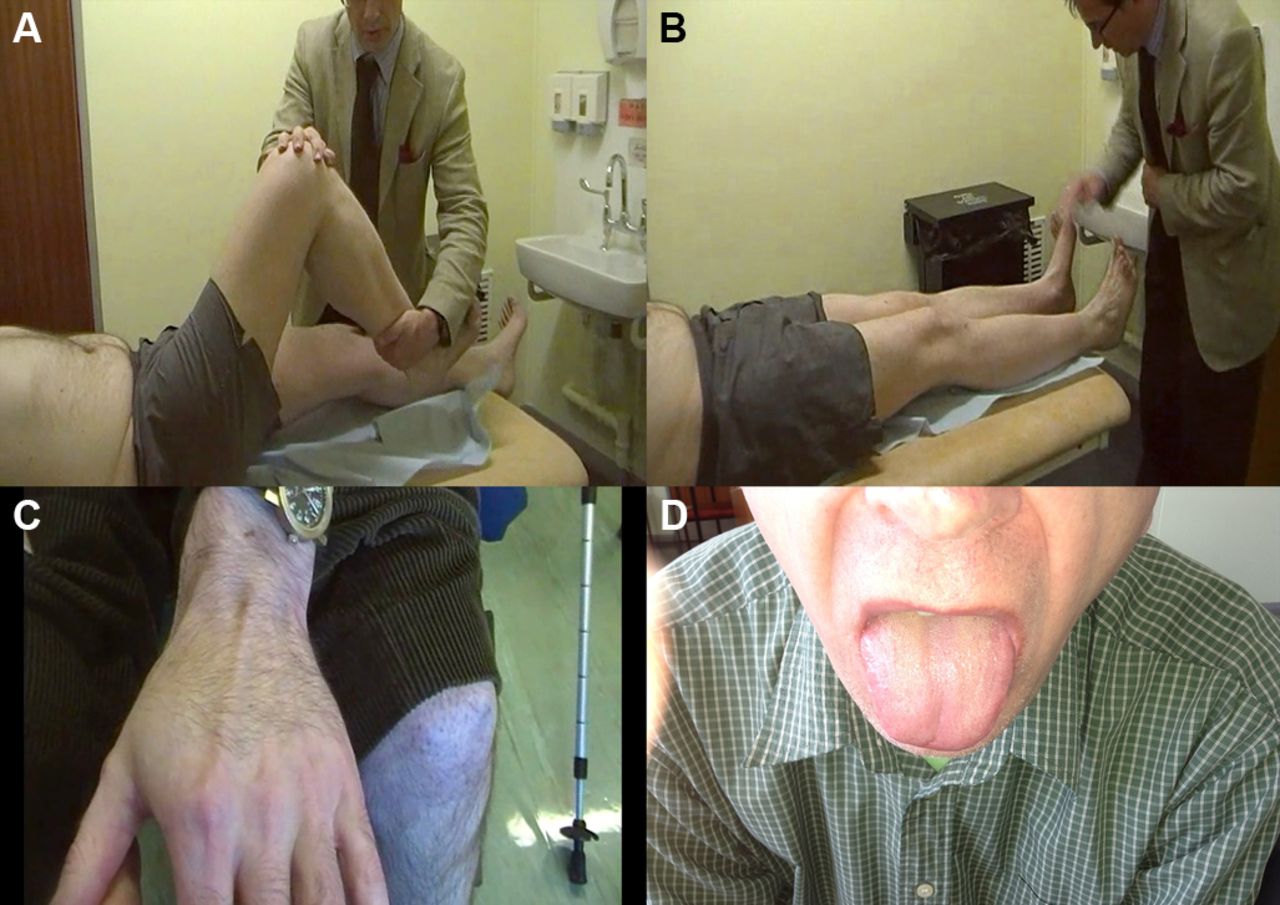
A patient with LGMD2I showing bulky calf muscles (A) with large and prominent extensor digitorum brevis and first dorsal interosseus (B and C, respectively) and macroglossia (D).

This man presented in his late 20s with proximal lower limb weakness and raised serum creatine kinase (3500 IU/L). He had asymmetric atrophy of his left medial calf (and left quadriceps) with genetically proven LGMD2L.
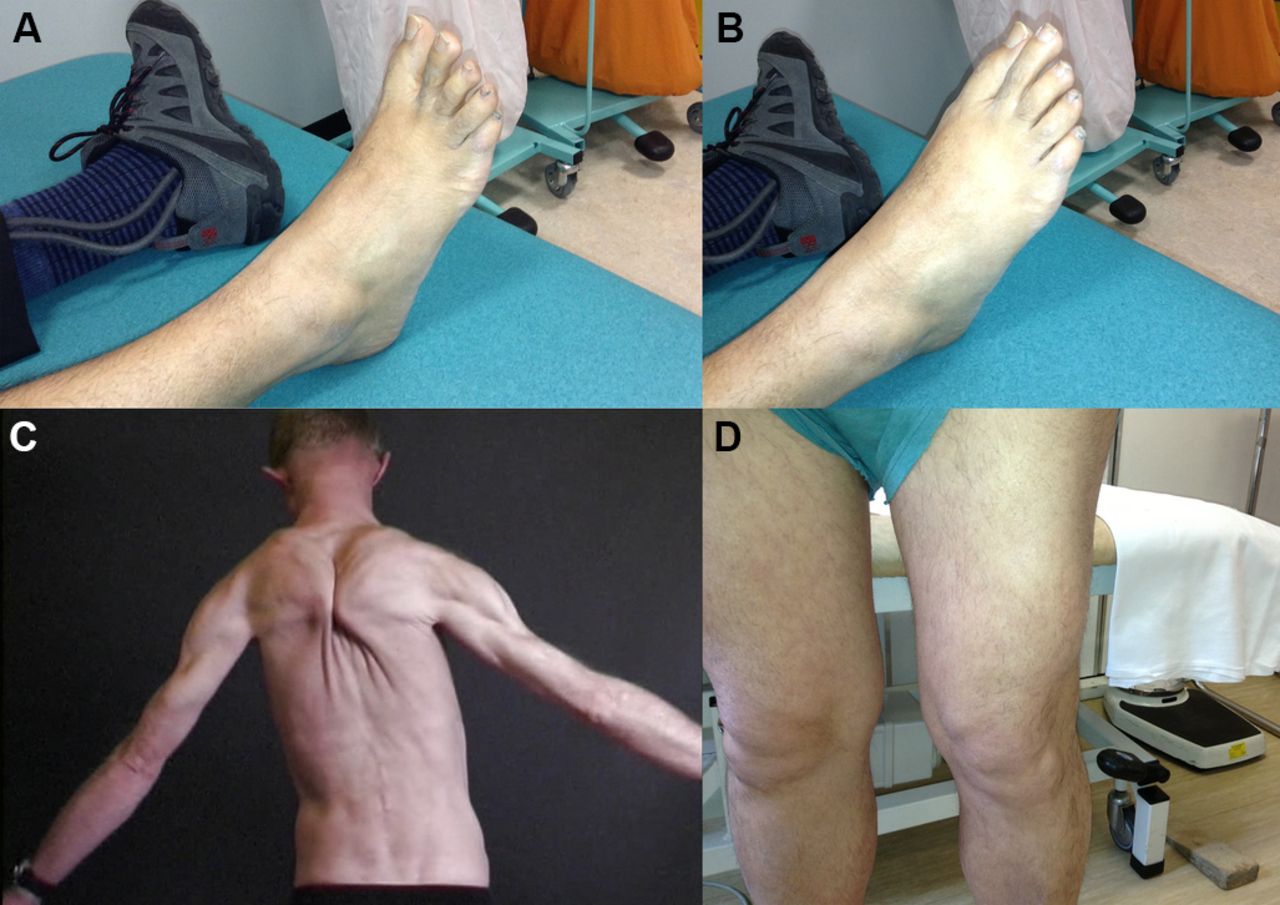
Muscle hypertrophy in various muscle dystrophies. (A) Selective hypertrophy of the lateral quadriceps of a patient with an alpha-sarcoglycanopathy. (B) Paraspinal muscle enlargement with calveolinopathy (LGMD1C, autosomal dominant). (C, D) A patient with facioscapulohumeral dystrophy who had prominent foot drop. At rest (C: note ankle foot orthosis on left foot) and when extending the toes (D) there is well-preserved right extensor digitorum brevis that contrasts with the atrophy that accompanies neurogenic causes of foot drop. The deltoid muscle can also be relatively enlarged in facioscapulohumeral dystrophy, the ‘poly-hill sign’.35
LGMD2I is an autosomal recessive condition with variable severity. Generally, the serum creatine kinase concentration is high (1000–5000 IU/L). There is calf pseudohypertrophy and sometimes macroglossia, a limb-girdle distribution of weakness, no contractures, often scapular winging and lower limb MR imaging shows fatty replacement in the posterior thigh (particularly biceps femoris) with early cardiac and ventilatory muscle involvement. A common mutation in the fukutin-related protein gene (C826A) accounts for 90% of cases.14
Sarcoglycanopathies (LGMD2C–F) occur less commonly in UK practice but are also characterised by calf pseudohypertrophy. Indeed, LGMD2G (telethoninopathy) causes calf hypertrophy in about half of affected individuals.14
A LGMD ‘genetic panel’ using next generation sequencing technology will probably play an increasing role in diagnosis. Certainly, genetic testing for Becker’s muscular dystrophy and LGMD2I can be justified before muscle biopsy in patients with limb-girdle weakness, raised serum creatine kinase and pseudohypertrophy. It is critical to remember that the differential diagnosis of limb-girdle weakness with pseudohypertrophy is not confined to Becker’s muscular dystrophy and LGMD but includes those neurogenic conditions already outlined, as well as other myopathies, for example, some metabolic conditions and myotonic dystrophy type 2 (see below). We need therefore to keep our wits about us before embarking down an ultimately futile battery of genetic tests; clinical acumen, electrophysiology and muscle biopsy remain essential to reach a diagnosis.21
Metabolic myopathies
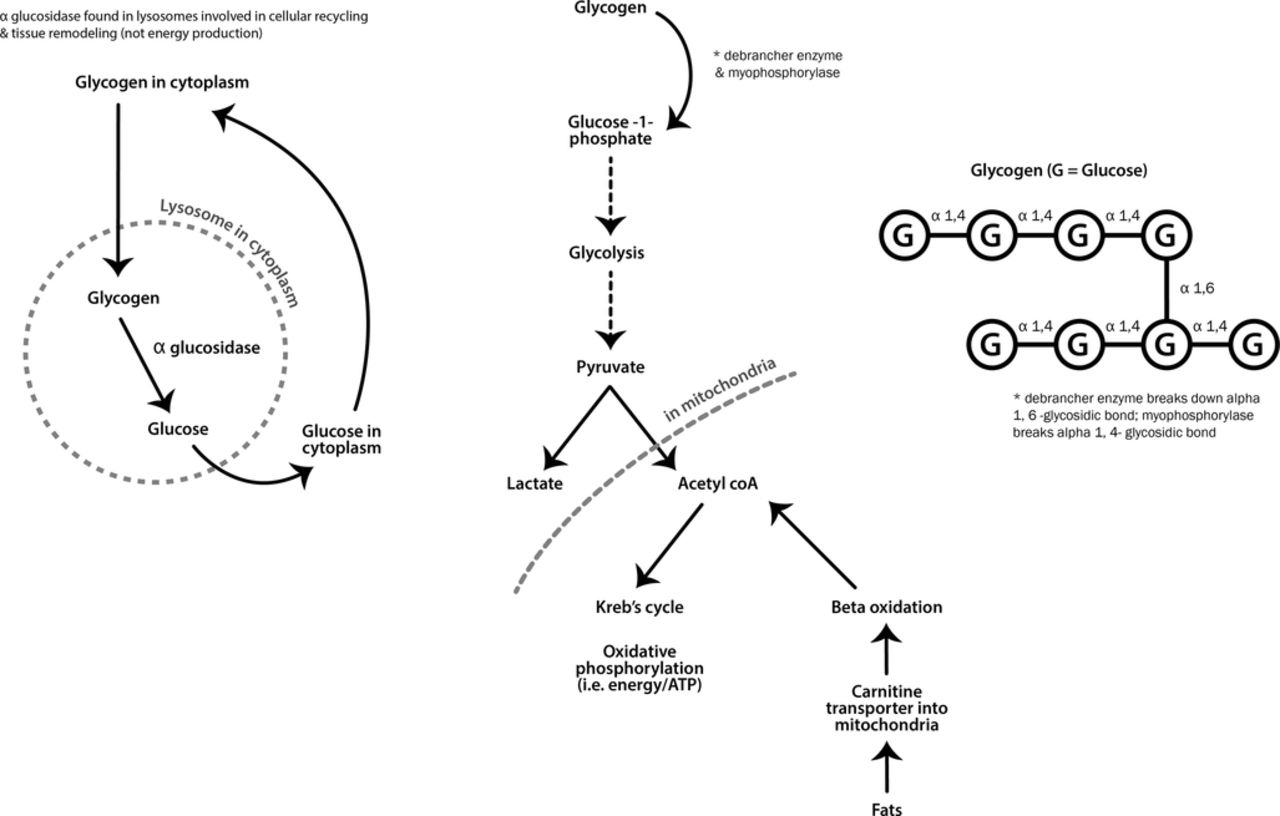
Metabolic myopathies generally present with exercise-induced symptoms when energy supply cannot keep up with demands. At the start of strenuous exercise, muscles use glycogen for fuel, whereas fat provides energy during longer low-intensity exercise (see figure 7).

Metabolic pathways for muscle energy production.
Some metabolic myopathies, however, present with predominately static, progressive weakness and muscle hypertrophy. This group includes Pompe’s disease (acid maltase deficiency, also known as acid α-glucosidase enzyme deficiency) and debrancher deficiency (Cori-Forbes disease) (see figure 7).
Pompe’s disease typically presents with lower limb weakness, particularly of hip flexion, often a hyperlordosis and marked Trendelenburg gait with mild scapular winging. It can be mistaken for a LGMD or Becker’s muscular dystrophy, since calf hypertrophy sometimes occurs. One-third of cases present with early ventilatory muscle weakness while patients are still ambulant. There are myotonic discharges on electromyography in paraspinal muscles. The serum creatine kinase can be normal and importantly muscle biopsy may not always be diagnostic. A misdiagnosis of spinal muscular atrophy is not uncommon. This emphasises the importance of performing a dried blood spot enzyme assay for acid maltase early in diagnostic workups to ensure that Pompe’s disease is not missed. This enzyme is not part of the glycolytic pathway, and so there is no exercise intolerance (see figures 7 and 8). There is now enzyme replacement available for acid maltase deficiency, which appears to stabilise ventilatory function and improve walking after 18 months follow-up.22
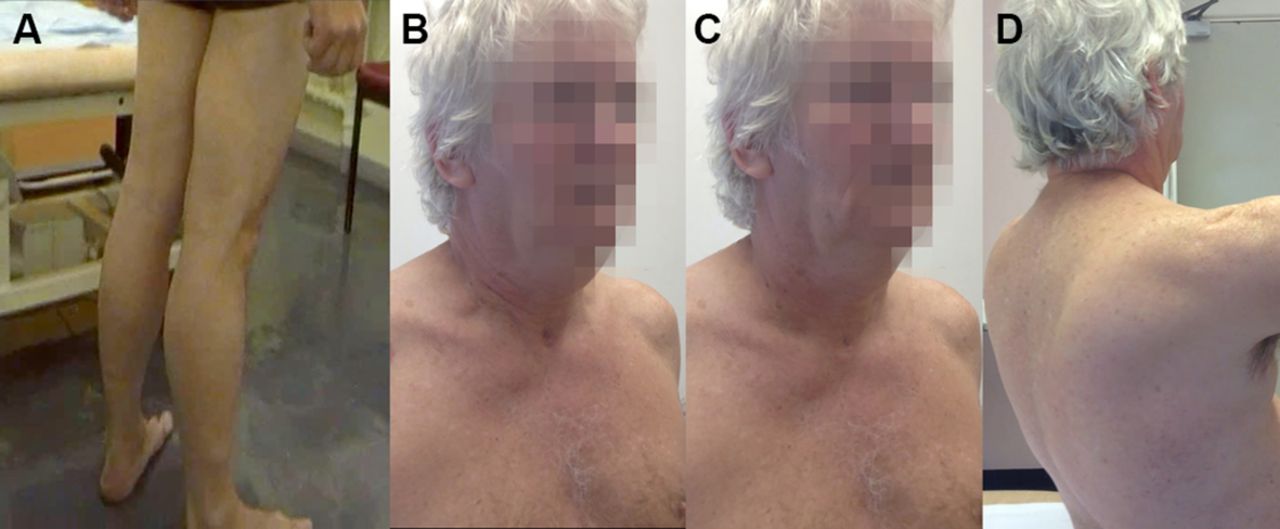
Adult form of acid maltase deficiency. (A) Relative preservation of calf bulk that can be more striking. There was no macroglossia, since this tends to occur in the infantile form characterised by enormous glycogen accumulation from the first few months of life. This man also had diaphragm weakness and in order to elevate his chest and breathe in, he used the muscles that suspended his rib cage from his neck, accessory muscles of ventilation such as sternocleidomastoids and scalene muscles: (B) Inspiration and (C) expiration. (D) He had a typical Trendelenburg gait, with his arms often held behind him for stability and a significant lumbar lordosis with mild scapular winging (figure 8D).
Debrancher enzyme is needed for the complete breakdown of glycogen. Myophosphorylase can still cleave terminal glucose residues from glycogen but without debranching enzyme, a limit dextrin remains (an abnormal glycogen with short outer chains) and accumulates in muscle, heart and liver (see figure 7). Patients with debrancher enzyme deficiency (Cori-Forbes disease) can therefore liberate some glucose from glycogen meaning that their exercise-induced symptoms are often not severe. Cori-Forbes disease tends to present with childhood hepatic problems and hypoglycaemia that often resolve around puberty; some patients then develop a slowly progressive weakness, occasionally with muscle hypertrophy (see figure 9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A patient with Cori-Forbes disease who had childhood hepatomegaly and hypoglycaemic episodes; despite a liver biopsy, there was no diagnosis. Later, he had a very physical job in the steelworks and gradually could not keep up with the physical demands. He attended clinic in his late 30s and was still walking, but he had proximal leg weakness and proximal and distal upper limb weakness. His serum creatine kinase was 2000–3000 IU/L. He had massive leg muscles but noticeable wasting of the small muscles in his hands. Electromyography appeared myopathic and nerve conduction showed normal sensory action potentials with normal MR imaging of the cervical spine. His diagnosis was confirmed genetically.
Cardiac involvement occurs in Cori-Forbes disease and regular cardiac assessment is vital. There is a blunted (or sometimes normal) exercise-induced lactate response in the aerobic forearm test, but the diagnosis is confirmed genetically.
Myotonia and muscle hypertrophy
The dystrophic myotonias
The most common myotonic disorder in a neurology clinic is, of course, myotonic dystrophy type 1 (DM1). It is the most common adult-onset muscular dystrophy with prevalence of 1 in 8000.23 It presents in several ways but classically in adult-onset cases there is facial weakness and ptosis, neck flexion weakness and early atrophy and weakness of the long finger flexors, leading to disabling loss of hand grip and foot drop. It is a progressive multisystem disorder.
Myotonic dystrophy type 2 (DM2) occurs less often. It can resemble a limb-girdle myopathy and 50% of patients have pseudohypertrophy of the calves. In general, it is less severe than DM1 (see box 1).
Myotonic dystrophy type 2 (DM2) case
A 65-year-old man presented with a 5-year history of progressive proximal lower limb weakness, such that he was now struggling to walk upstairs. He reported myalgia particularly in his thighs. He had also noticed loss of grip strength when fishing, confirmed by finding weakness of long finger flexors. The gestalt was that this looked like inclusion body myositis but he had large calves and also a history of abnormal liver tests investigated in his 20s and cataracts removed in his 40s. His father and his daughter also had cataracts. Genetic testing for DM2 was positive. He had no clinical or electrophysiological evidence of myotonia (even with the benefit of knowing the diagnosis). Fewer than 50% have clinical myotonia and not infrequently it is absent on electromyography. DM2 tends to present later in life with myalgia and proximal weakness; a few patients can have weak finger flexors too and about half have calf hypertrophy. It is a multisystem disorder although frequently less severe than DM1. Cataracts are a clue and, importantly, cardiac conduction defects can range from absent to severe.
The non-dystrophic myotonias
True muscle hypertrophy develops in some of the non-dystrophic myotonias, a direct result of muscle overactivity. These enlarged muscles do not have paradoxical weakness and often exhibit increased strength.
The non-dystrophic myotonias include disorders that arise from a chloride ion disorder (CLCN1 gene, CLC11 protein), namely myotonia congenita, and from sodium channel mutations in the SCN4A gene (NaV1.4 protein), paramyotonia congenita and sodium channel myotonia.14
Patients with myotonia complain of muscle stiffness. In myotonia congenita, the stiffness improves with activity (warm-up). The patient getting up from the chair in the waiting room when the clinic is running an hour behind walks stiffly and slowly like a rusted tin person, but by the time they reach the consultation room they are moving much more easily; potentially this can resemble an extrapyramidal disorder or Lambert-Eaton myasthenic syndrome.
Patients may have a ‘Herculean’ appearance, particularly in the calves and buttocks. The masseters are often hypertrophied causing a distinct appearance that contrasts with the atrophy of the temporalis muscle in myotonic dystrophy type 1. There is usually action and percussion myotonia.
The sodium channelopathies often show myotonia that is very cold sensitive, worsens with activity (paradoxical myotonia) and involves the eyelids and face. There may be episodic weakness in paramyotonia congenita but not in sodium channel myotonia. Sometimes the myotonia in sodium channel myotonia can exhibit a warm-up phenomenon.
These conditions are ultimately diagnosed genetically but, as well as careful clinical assessment, several electrophysiological studies can help including electromyography and the short and long exercise tests.14
Endocrine conditions
Hypothyroidism often causes myalgia, cramps and raised serum creatine kinase and characteristic delay of relaxation after eliciting tendon reflexes (Woltman’s sign). Sometimes, there is an electrophysiologically silent ‘mounding’ of muscle following percussion (myoedema). Occasionally, in long-standing untreated hypothyroidism, there is muscle hypertrophy and painful muscle spasms (Hoffman’s syndrome).
For completeness, focal enlargement of the extraocular muscles may accompany dysthyroid eye disease. It is caused by oedema, lymphocytic infiltration and proliferation of fibroblasts and most commonly occurs in Graves’ disease and less frequently in Hashimoto’s thyroiditis. The most likely cause is an antigen shared between the orbital tissue and thyroid gland.14 In dysthyroid eye disease, imaging shows spindle-shaped muscle enlargement (most frequently inferior rectus) that spares the tendon.
Other endocrinopathies, for example, acromegaly, can also cause mild proximal weakness with muscle hypertrophy too.
Parasitic muscle conditions
Parasitic myositis, most commonly resulting from infection with cysticercosis, toxoplasmosis or trichinosis, occasionally appears in UK neurology practice. Typically, patients have travelled to, or been resident in an endemic area, and have a suggestive clinical picture often including an eosinophilia. The eggs from the cestode Taenia solium, the pork tapeworm, are activated by gastric juices and the larvae penetrate the gastrointestinal wall disseminating to the central nervous system, subcutaneous tissues, eye and skeletal muscle through the bloodstream. Cysticercosis therefore represents an infection by the larval stage of T. solium. Skeletal muscle is involved in 75% of cases of neurocysticercosis and although this is often asymptomatic, it can be characterised by pseudohypertrophy, often but not exclusively of the calf muscles. The skeletal muscle pathology reveals inflammation both around cysts and elsewhere in the muscle, with myofibre swelling.24
Amyloid and sarcoid myopathies
Skeletal muscle pseudohypertrophy may also develop in amyloidosis, sometimes as the presenting complaint. Associated clinical clues can include the ‘shoulder pad’ sign—shoulder enlargement resulting from amyloid deposition in synovial and periarticular tissue—and the ‘raccoon’ sign of periorbital ecchymoses; muscle amyloid disease can also cause claudication.25 26 It reminds us that when suspecting a neuropathy or myopathy, we should have a low threshold for undertaking protein electrophoresis and potentially also for immunofixation and serum-free light chain assay.
Muscle involvement in sarcoidosis can cause an acute, subacute or more commonly a chronic weakness, subcutaneous nodules and myalgia and occasionally pseudohypertrophy as well as atrophy. Non-caseating granulomas are found in 50% or more of muscle biopsies from patients with sarcoidosis who have no clinical evidence of muscle disease. Thus, a generous muscle biopsy might be useful when suspecting neurosarcoidosis, even without any systemic clinical abnormalities.27–29
Key points
Recognising muscle enlargement provides a useful clinical pointer to the diagnosis of neuromuscular weakness.
Calf hypertrophy develops in a wide range of myopathic conditions; clinical acumen, electrophysiology and possibly muscle biopsy remain important to avoid a potentially fruitless genetic quest.
Neurogenic disorders can cause muscle hypertrophy and particularly autosomal recessive spinal muscular atrophy; several clinical clues can help to distinguish these from myopathic disorders.
Do not assume a neurological or myopathic cause for focal muscle enlargement—beware venous thrombosis or sarcoma in particular.
References
Footnotes
Contributors This is solely my own work.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Wojtek Rakowicz, Hampshire, UK.
Other content recommended for you
- Muscle diseases: mimics and chameleons
- Muscle disease
- Diagnosis and management of the limb girdle muscular dystrophies
- Making sense of the clinical spectrum of limb girdle muscular dystrophies
- Muscle biopsy: what and why and when?
- Current state of cardiac troponin testing in Duchenne muscular dystrophy cardiomyopathy: review and recommendations from the Parent Project Muscular Dystrophy expert panel
- Effectiveness of conservative non-pharmacological interventions in people with muscular dystrophies: a systematic review and meta-analysis
- Approaches to genetic diagnosis in neuromuscular conditions in the era of next generation sequencing
- Respiratory involvement in inherited primary muscle conditions
- Diagnosis and new treatments in muscular dystrophies