Article Text

Abstract
General neurologists and stroke specialists are regularly referred cases of visual disturbance by general practitioners, emergency doctors and even ophthalmologists. Particularly when the referral comes from ophthalmologists, our assessment tends to focus on the optic nerve; however, retinal conditions may mimic optic neuropathy and are easily missed. Their diagnosis requires specific investigations that are rarely available in a neurology clinic. This article focuses on how a general neurologist can identify retinal problems from the clinical assessment and how to proceed with initial investigations. The following cases were all referred to a consultant neurologist (GTP) from ophthalmology services as optic neuropathies or other neurological disorders. Part A of the summary describes the presentation and findings in the neurology clinic; part B describes the subsequent specialist assessment in the neuro-ophthalmology/eye clinic.
- neuro-ophthalmology,
- Retinopathy
- Optic neuritis
- white dot syndromes
- AZOOR
Statistics from Altmetric.com
Introduction
The following cases were all referred to a consultant neurologist (GTP) from ophthalmology services as optic neuropathies or other neurological disorders. Part A of the summary describes the presentation and findings in the neurology clinic; part B describes the subsequent specialist assessment in the neuro-ophthalmology/eye clinic.
Case 1
Part A
A 34-year-old woman presented with blurred vision for 5 days. When viewing with the right eye, there were multiple bright white spots in her vision and red colours looked ‘washed out’. When she was reading, the area around her point of fixation would ‘grey out’.
She had a history of coeliac disease, Graves’ disease and depression. Her father had a history of glaucoma. Her medications were regular B12 injections and citalopram. She was referred to neurology with suspected optic neuritis.
What is your differential diagnosis? What else would you ask?
The patient has photopsia and is describing phosphenes (bright lights). These can originate in the retina, the optic nerve or the cortex.
Retinal phosphenes are more visible in the dark. They are commonly due to vitreoretinal traction, which can cause vitreous detachment (usually benign) or at worst can result in retinal detachment (this would be visible on funduscopy and would not cause red desaturation). Phosphenes can also be caused by retinal pathology such as inflammation or infection (retinitis). If the macula is affected, there may be red desaturation.
Optic nerve phosphenes occur occasionally in acute optic neuritis. They are more visible in the dark and may occur only on eye movement. There is very frequently pain on moving the eyes—unless the inflammation is intracranial—and there may be a swollen disc (although most often the inflammation is retrobulbar), a relative afferent pupillary defect and red desaturation that often occurs before visual acuity loss.
Cortical phosphenes most commonly occur in migraine; occasionally they are due to occipital seizures. Neurologists will be familiar with their respective features. They are homonymous and unaffected by ambient light levels.
On direct questioning, the patient said the bright spots were more visible in the dark. They were constantly present and unrelated to eye movement. There was no pain. The left eye was unaffected. There was no preceding illness.
Remember the retina when…
There are phosphenes (flickering and shimmering) more visible in the dark.
There is distortion of images (straight lines become wavy, objects look a different size compared between the two eyes).
There is nyctalopia or ‘night blindness’, that is, reduced vision in dim light (rod dysfunction, which can be receptoral or postreceptoral).
The vision deteriorates in bright light (cone dysfunction, which can be receptoral or postreceptoral).
There is slower recovery in the affected eye after exposure to bright light.
Blind spot enlargement is not explained by disc swelling.
Scotomata are irregular, patchy or ring shaped, not respecting the vertical or horizontal meridians (known as geographic scotomas).
…think twice when diagnosing optic neuropathy…
Abnormalities of colour vision and photophobia may be retinal.
Disorders of the peripapillary retina may be accompanied by mild disc swelling and a relative afferent pupillary defect.13
On examination, acuities were right eye 6/18, improving to 6/12 with pinhole, and left eye 6/5. Ishihara right eye 15/17 and left eye 17/17. There was paracentral red desaturation in the right eye, with a patchy scotoma to confrontation. On viewing the Amsler grid (see box 2) with the right eye, there was patchy ‘greying out’, but no distortion. There was no relative afferent pupillary defect, and the optic discs looked healthy.
The Amsler grid
A grid of horizontal and vertical lines used regularly in eye clinics to assess the central visual field. It helps to identify retinal and particularly macular problems by detecting distortion and scotomata. It is most frequently used to monitor age-related macular degeneration.
The patient looks at the small dot in the centre of the grid. Patients with macular disease may see wavy lines or some lines may be missing. Each eye is examined separately.
The original Amsler grid contained white lines on a black background, but today Amsler grids often have black lines on a white background. It is unclear whether one style is better than the other (http://amslergrid.org, n.d.).14 Amsler grids come in notepad form and are an easy addition to a neurology toolkit.
These features indicate that there is a retinal problem requiring closer examination: phosphenes are more visible in the dark but unrelated to eye movements, patchy scotomata and no relative afferent pupillary defect, disc swelling or pain. The red desaturation could indicate optic nerve involvement but can also occur with dysfunction of the cone photoreceptors or postreceptoral cells in the macula.
Part B
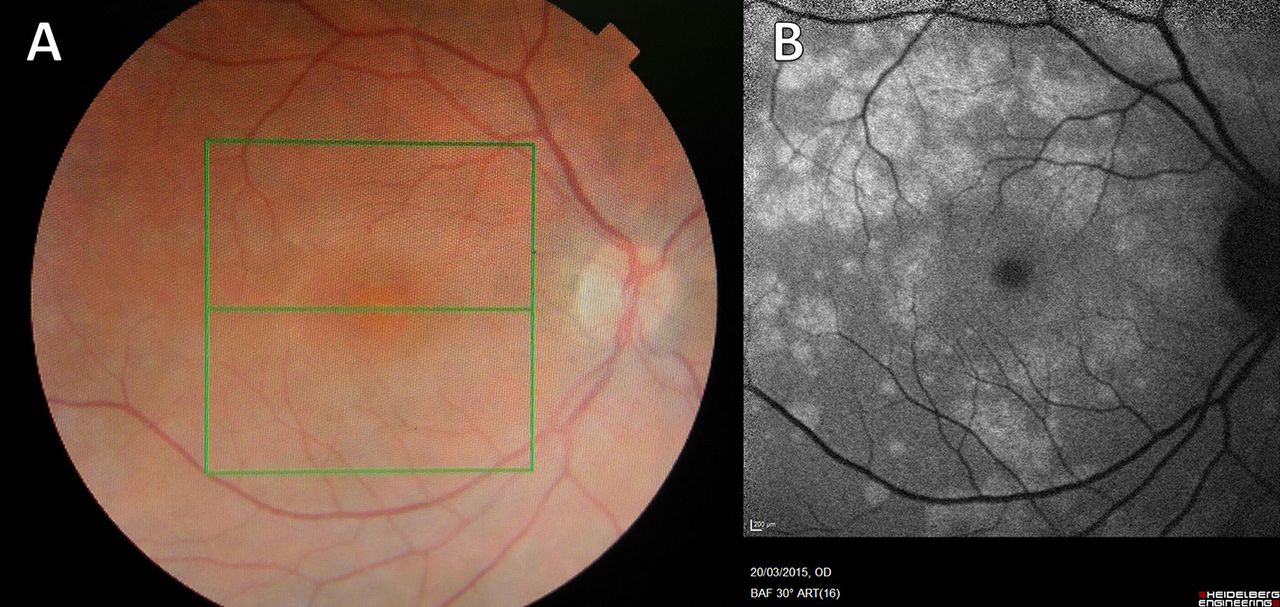
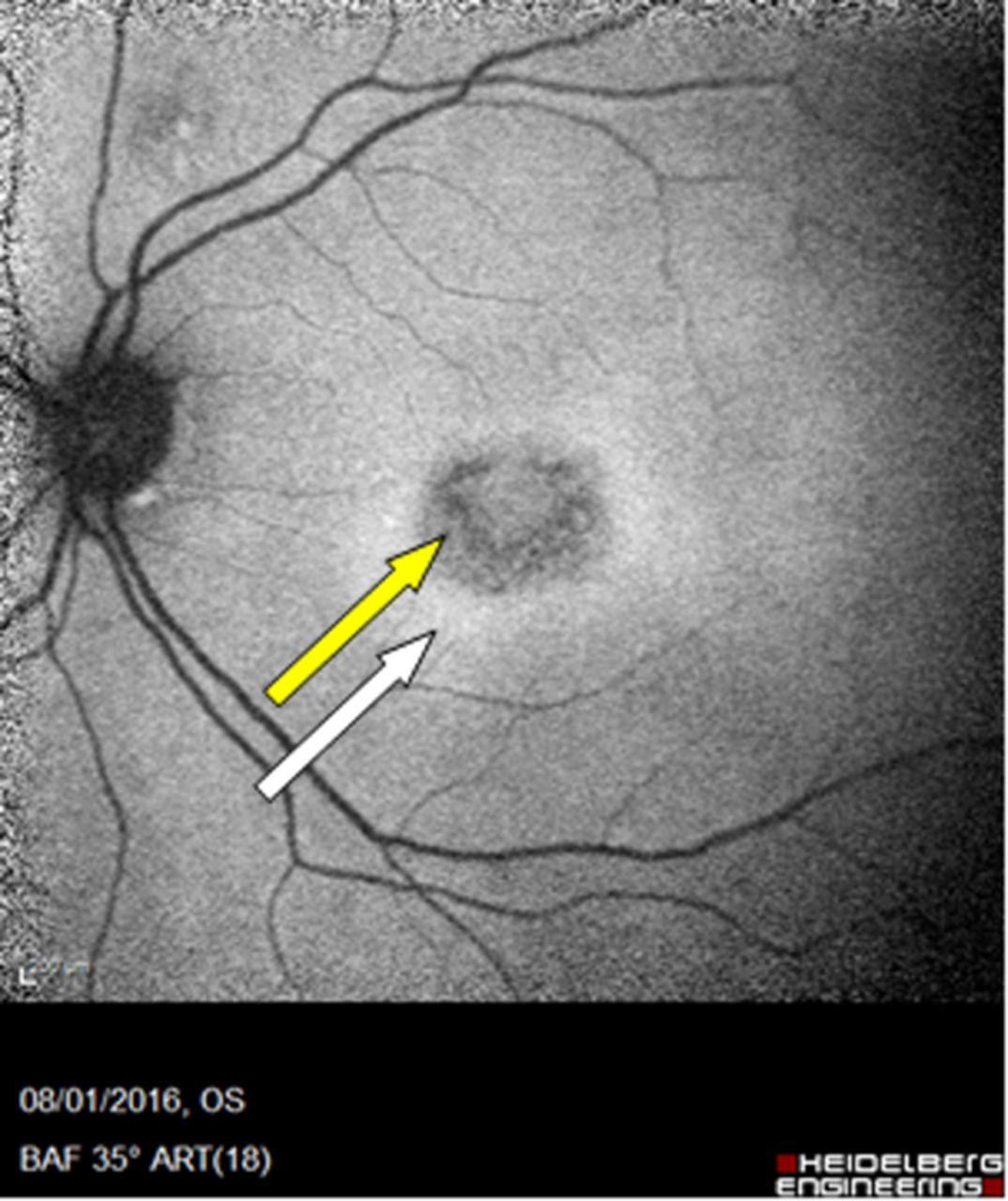
On slit lamp examination, there was no inflammation in the vitreous humour (vitritis). There were small white retinal patches and dots around the disc, which were more clearly seen on fundal photographs (see figure 1).
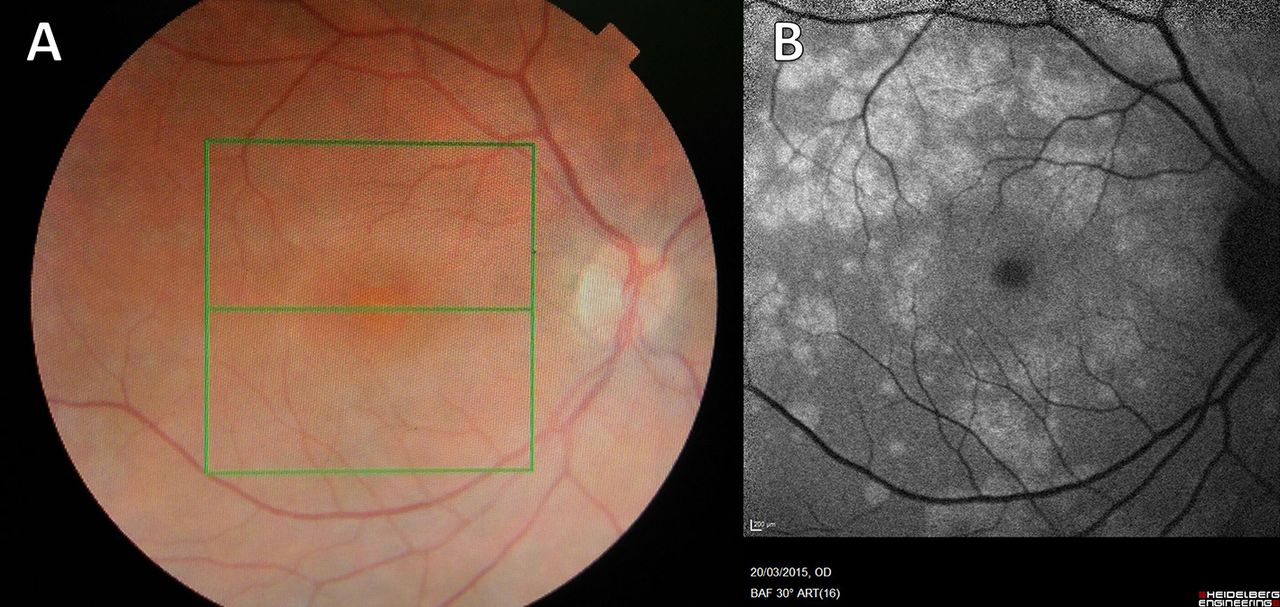
Fundal photograph (A) and autofluorescence (B) (Case 1). (A) There are multiple small, white or yellowish dots on the retina that may be confluent. They may be most dense around the macula. This appearance is typical of Multiple Evanescent White Dot Syndrome, a chorioretinopathy of the outer photoreceptor layer. Granularity at the fovea, not visible here, is pathognomic. The changes here are subtle but much more obvious using autofluorescence imaging. (B) Autofluorescence highlights areas of altered metabolism, and the optic disc and vessels appear dark. The white dots are clearly seen to coalesce around the vascular arcades and there is a density of white dots around the macula.
On optical coherence tomography (see box 3; see figure 2) the white dots were seen to represent areas of outer retinal disruption involving the photoreceptor layers and underlying retinal pigment epithelium. Autofluorescence imaging (see box 3) showed the white dots clearly, with a maximal density around the macula (see figure 1).

OCT right eye (case 1) at presentation. On this cross-sectional OCT, which is centred on the macula, disruption of the retinal pigment epithelial layer is seen (yellow arrows). OCT, optical coherence tomography.
Blood tests to screen for vasculitis, sarcoid and syphilis were normal or negative.
Diagnosis: multiple evanescent white dot syndrome (MEWDS)
MEWDS, first described in 1984, is a chorioretinopathy affecting the photoreceptors and causing changes in the choroid and retinal pigment epithelium.1 It typically affects young myopic women and in 30%–50% is preceded by a viral illness. Patients present with a paracentral scotoma or enlarged blind spot, photopsia and dyschromatopsia. There may be a relative afferent pupillary defect and mildly swollen disc, and there may be cells seen in the vitreous. MEWDS is usually unilateral, while most of the other white dot syndromes are bilateral. A full recovery is expected within weeks to months; our patient recovered fully after 3 months.
White dot syndrome has a wide differential diagnosis, many of which have additional features (see table 1 and figure 3). Patients need careful screening for other infective and inflammatory conditions affecting the retina (eg, sarcoid and syphilis). Ongoing management is by retinal specialists to confirm the diagnosis and to follow the patient in case of complications, which include bilateral and recurrent MEWDS and choroidal neovascularisation or scarring.
Differential diagnosis of white dot syndromes

Retinal layers as seen on optical coherence tomography.
Case 2
Part A
A 34-year-old man presented with an area of persistently blurred vision in his right inferotemporal visual field. It was described as flickering, ‘like lightning’ and had been present for 8 weeks. There was no history of headaches and he had been well in the past. He was referred to neurology with suspected optic neuritis.
On examination, visual acuities were right eye 6/4 left eye 6/5 with glasses. Ishihara plates were 17/17 bilaterally. Visual fields to confrontation showed an enlarged blind spot on the right. There was no relative afferent pupillary defect. Optic discs appeared healthy.
What is the differential diagnosis?
Patients with an enlarged blind spot typically have disc swelling; this causes displacement of the peripapillary retina and thus an enlarged blind spot. In the absence of marked disc swelling, patients need a careful retinal examination to look for chorioretinitis. If there is evidence of chorioretinitis, the patient probably has one of the retinal conditions described in table 1. If the retinal appearances are normal, or there are only mild peripapillary changes, a diagnosis of acute idiopathic big blind spot syndrome (AIBBSS) is made.
Part B
Undilated slit lamp examination found no abnormality; however, when the pupil was dilated, a variation in colour of the peripapillary retina was visible.
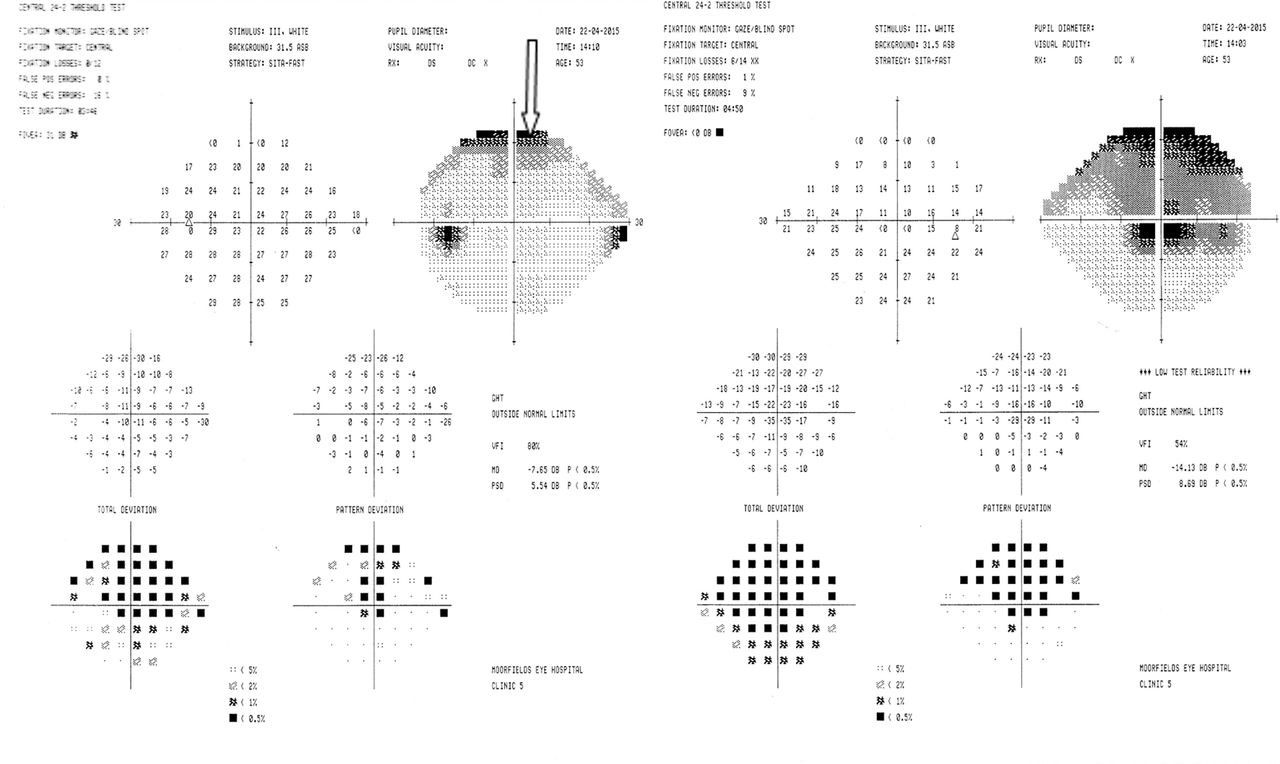
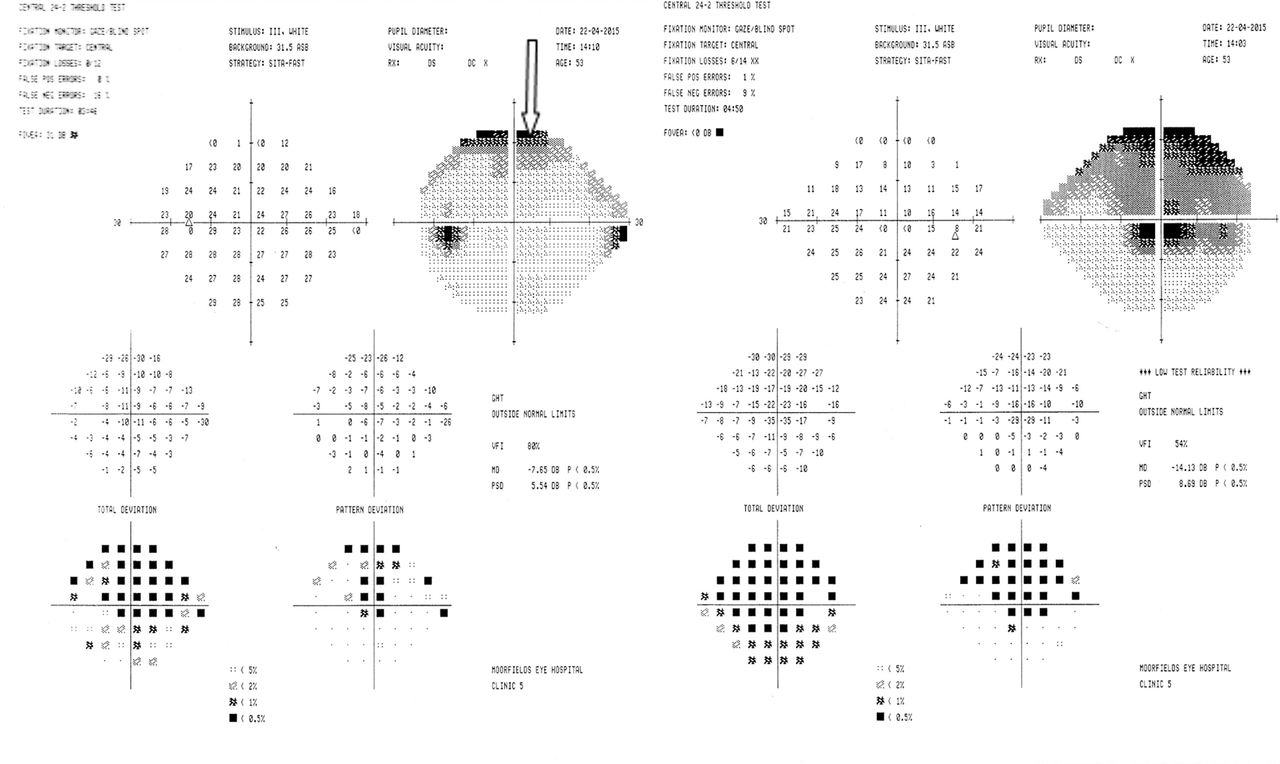
Humphrey field analyser visual field examination confirmed the presence of an enlarged blind spot on the right (figure 4).

Humphrey field analyser (case 2). Fields clearly demonstrate an enlarged blind spot in the right eye visual field and a normal blind spot on the left. In the absence of disc swelling, this suggests a retinal problem.
Autofluorescence imaging showed the abnormality in the peripapillary retina (see figure 5) and optical coherence tomography showed thinning of the peripapillary retina (not shown).

Autofluorescence of the right eye (case 2). There is hyperautofluorescence, indicating altered metabolism, in a ring around the right disc. The rest of the retina is normal. This appearance is typical of acute idiopathic big blind spot syndrome.
Diagnosis: AIBBSS
AIBBSS is a condition affecting the peripapillary retina. It is due to dysfunction of the outer segment of the retinal photoreceptor layer. Like MEWDS, it has a predilection for myopic females. Patients experience photopsia, blurring and dyschromatopsia. There may be a relative afferent pupillary defect. The fellow eye is normal but with time sometimes becomes affected. Visual symptoms usually resolve, but the enlarged blind spot tends to remain. There may be some link with MEWDS, as this can occur without visible white dots; however, unlike MEWDS, there are no cells in the vitreous, and the blind spot remains enlarged.2 There may also be overlap with other white dot syndromes (table 1).
Case 3
Part A
A 68-year-old woman attended the stroke clinic, several months after visiting an ophthalmic emergency room with transient monocular visual loss. She described a black curtain coming down until she experienced complete, painless loss of vision in the right eye for about 90 minutes. The vision recovered gradually. However, she complained that there was one abnormal area remaining. On close questioning at a subsequent visit, she eventually admitted to blurred vision in both eyes. It was worst on the right, in the centre of her vision, and made reading difficult.
She had a complex medical history. There was a history of migraine with visual aura and possible seizures, treated for some years with phenytoin, which was now stopped. She had undergone eyelid surgery bilaterally for dysthyroid eye disease. She had been treated with radioiodine and subsequently developed hypothyroidism. She had diagnoses of Sjögren’s syndrome, hypertension, irritable bowel syndrome and fibromyalgia. She had been under regular ophthalmological follow-up for her dysthyroid eye disease and Sjögren’s syndrome. There was a family history of multiple sclerosis in her sister and niece.
Visual acuities were right eye 6/12 and left eye 6/9 with glasses. Ishihara plates were ‘none read’ with the right eye, and only the first (control) plate was readable with the left eye (she knew her colour vision had been normal 2 years before, as she had been tested at the dysthyroid eye clinic). There was no relative afferent pupillary defect. Her discs were small and crowded but appeared healthy. Visual fields to confrontation and funduscopy were normal. Neurological examination of the limbs was normal.
Magnetic resonance (MR) imaging of brain, 24-hour ECG and carotid Doppler were normal. She was referred to neuro-ophthalmology as a case of vascular disease with persistent visual loss of unknown cause.
What is your differential diagnosis?
Which factors in her history may be relevant?
Which further tests may help?
There is asymmetric loss of central vision bilaterally. There is no relative afferent pupillary defect, optic atrophy or pain, but colour vision is disproportionately affected with preserved acuities. Disproportionate loss of colour vision is a sensitive indicator of optic nerve pathology but could also implicate the cone photoreceptors at the macula, particularly as this case is bilateral and progressive.
The episode of transient visual loss was probably vascular in origin but led to an initial misdiagnosis of what was thought to be some form of residual loss.
The differential diagnosis is
bilateral optic neuropathy with central field loss
chiasmal lesion causing bilateral optic nerve dysfunction, but this does not fit with the visual field abnormality
bilateral maculopathy, slowly progressive and possibly hereditary; the drug history is essential here to look at toxic causes.
Further tests must be directed at looking more closely at the macula.
Part B
Slit lamp examination showed subtle retinal pigment epithelial layer changes at both maculae, particularly in the perifoveal area.
Optical coherence tomography showed bilateral foveal atrophy, with an island of preserved retina in the left fovea, known as a ‘bull’s eye maculopathy’ (figure 6).

OCT left eye (case 3). OCT showed bilateral foveal atrophy (white arrows), with an island of preserved retina in the left fovea (yellow arrow; only left eye shown); this is known as a ‘bull’s eye’ appearance and can occur in a variety of toxic, hereditary and degenerative maculopathies. OCT, optical coherence tomography.
Autofluorescence imaging showed hyperautofluorescence at the fovea (see figure 7).

Autofluorescence left eye (case 3). There were bilateral concentric bands of hyperautofluorescence and hypoautofluorescence (white and yellow arrows, respectively) at the fovea demonstrating altered metabolism.
Electroretinogram showed severe macular dysfunction bilaterally with no evidence of rod or cone dysfunction (ie, the dysfunction was postreceptoral).
It finally emerged, after a detailed drug history, that she had been taking hydroxychloroquine for 17 years. Hydroxychloroquine was stopped, and her visual acuity remained stable thereafter.
Diagnosis: hydroxychloroquine toxicity
Chloroquine and hydroxychloroquine are the best known retinotoxic drugs. Hydroxychloroquine is an antimalarial used to treat several rheumatological conditions. Those with short or slim stature require dose adjustment to their body weight. The incidence of retinal toxicity increases sharply after around 5–7 years of continuous use, and studies at 15 years have put the incidence at 4%. The presentation is with difficulty reading and loss of colour vision, with very subtle scotomata seen initially. Retinal changes include stippling of the macula, loss of the foveal light reflex and changes in pigmentation; the characteristic bull’s eye maculopathy only develops at an advanced stage. The changes are largely irreversible despite stopping the medication. Revised guidelines from the American Academy of Ophthalmology state that all patients must be screened at baseline for macular disease (a contraindication to treatment) and then monitored annually from at least 5 years (sometimes earlier) using, in addition to automated fields with detailed examination of the central 10° (10–2 protocol on Humphrey), at least one new objective measure of retinal function: multifocal electroretinogram, spectral domain optical coherence tomography or fundus autofluorescence. Where this is not possible, even subtle 10–2 field changes should be an indication for evaluation by objective testing.3
Other toxic maculopathies: tamoxifen, canthaxanthin and methoxyflurane cause toxic crystalline maculopathy in which the macula is surrounded by tiny glistening crystals; phenothiazines can cause a retinopathy in which patients present with blurring or browning of the vision and the macula may show pigment clumping.
The main differential diagnosis of toxic maculopathy is hereditary maculopathy, which also causes insidious loss of central vision. Hereditary maculopathies are usually diagnosed in late childhood but can present as late as the fifth decade. There are visible abnormalities of the retina.
Best’s disease is characterised by bilateral, well defined, round, yellow lesions that look like egg yolks. Despite this appearance, the vision is usually preserved until the fifth decade of life.
Adult vitelliform dystrophy has a similar appearance to Best’s disease but the ‘egg yolk’ lesions tend to be smaller. The onset is in the fourth to fifth decade.
Stargardt disease is the most common juvenile macular dystrophy. The fovea has a beaten bronze appearance surrounded by yellow flecks and later becomes a bull’s eye maculopathy. Fundus flavimaculatus is the adult onset form of Stargardt disease, presenting in the fifth to sixth decade.
Case 4
Part A
A 51-year-old woman, previously seen at another centre, presented for a second opinion with a 4-year history of central clouding in her vision, ‘like looking through condensation on a window’. The problem started with difficulty seeing road signs while driving and then in recognising faces at a distance. Initially, the problem appeared to be only in the right eye but later started to involve the left eye as well. There were gaps in her vision around fixation and some wavy distortion while reading. There were flashes of light in the centre of her visual field on occasion when closing the right eye. She had noticed that it took her longer than normal to adapt to darkness, and she found dimly lit environments problematic.
There was no medical or drug history. She was a non-smoker.
Visual acuities were right eye 6/12 and left eye 6/6 with pinhole. On Ishihara testing, she was a little slow reading with the right eye but accurate. There was some distortion of lines on the Amsler grid (see figure 8). There was no relative afferent pupillary defect and funduscopy was normal. The diagnosis at the referring hospital had been bilateral visual loss of unknown cause, possibly non-organic.
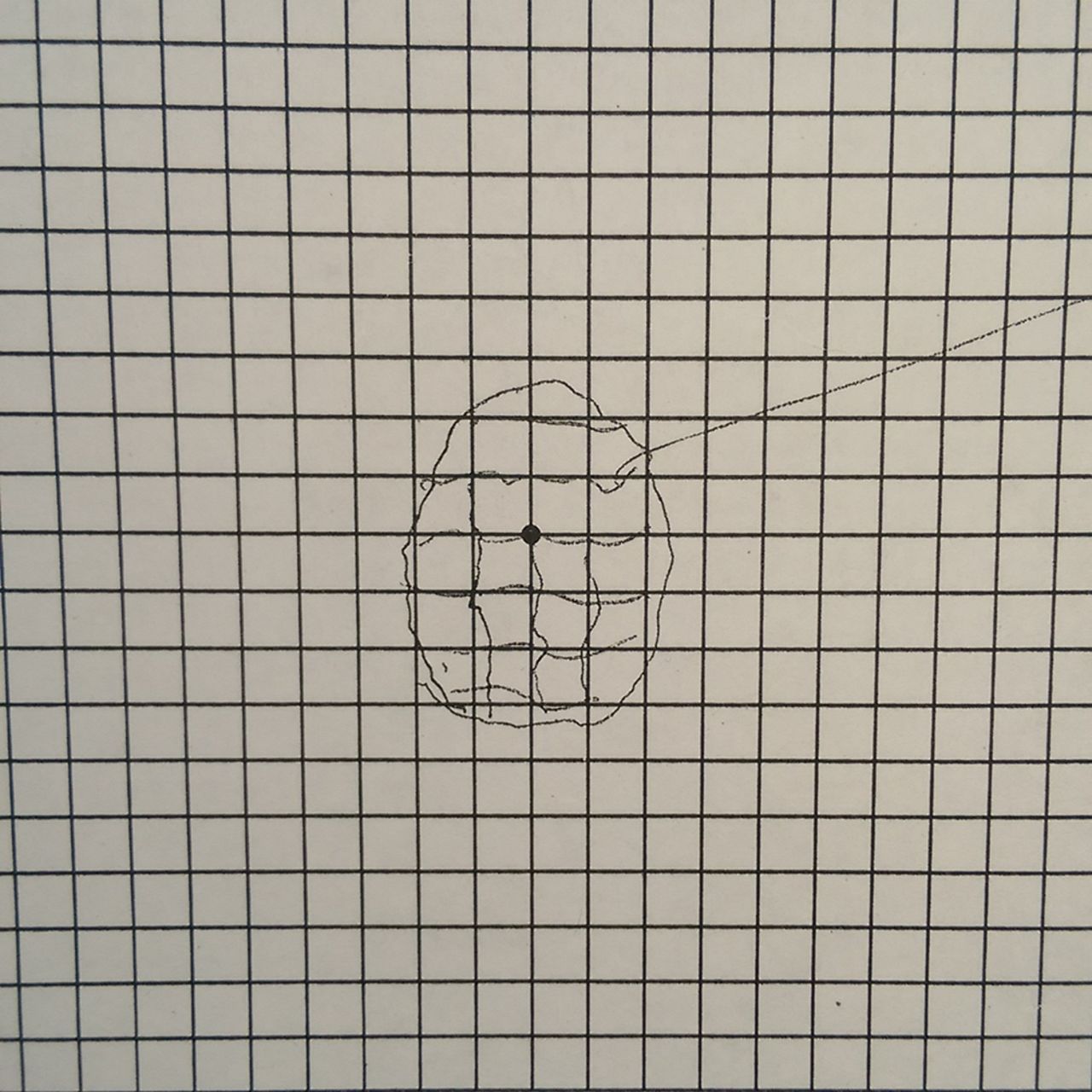
Amsler grid (case 4). The patient fixates the black spot and describes or draws what he or she sees on to the grid. This demonstrates distortion of the lines in central vision (see box 2).
Where is the lesion?
The difficulty with signs and faces implicates central vision at the macula, and the process is bilateral. The distortion, photopsia and difficulty in dim light all point to a retinal problem. In this case, colour vision was preserved, suggesting dysfunction of the paramacular rod system. The retina has a duplex function related to scotopic vision (rod function) and photopic vision (cone function). Any marked difference in vision at high and low light levels likely indicates retinopathy, because in the optic nerve, the photic and scotopic information is multiplexed in all axons. In optic nerve disease vision may vary with change in light level but never dramatically.
Part B
Slit lamp examination suggested a dim foveal light reflex from the right macula. There was a normal anterior chamber.
Optical coherence tomography showed no retinal abnormality.
What would be the most useful next test?
On electrodiagnostic studies, the electroretinogram showed delay in the maximal b-wave component, suggesting dysfunction of the inner retinal layers. Visual evoked potential amplitudes fell within the normal range.
Diagnosis: cryptic maculopathy
This concept of ‘cryptic’ maculopathy reflects the limitations of imaging in retinal disorders, in cases where retinal dysfunction (revealed by symptoms and electrophysiological abnormalities) does not have a structural correlate on imaging. In some cases electrophysiology alone is abnormal.
Case 5
A 54-year-old man presented to Moorfields Eye Hospital, London, having noticed poor vision in his right eye 6 days after a motorcycle accident, when he happened to cover the left eye. The accident had resulted in head injury with loss of consciousness. He had been admitted to hospital overnight and discharged next day. He was referred as a traumatic optic neuropathy, directly to the neuro-ophthalmology clinic.
Visual acuities were right eye 2/60 and left eye 6/5. There was red desaturation on the right. There was a right-sided relative afferent pupillary defect; his optic discs appeared healthy, and at his initial assessment, there was some retinal swelling. Visual field testing to confrontation found a central scotoma on the right, confirmed with visual fields (see figure 9).
Humphrey field analyser (case 5). There is a dense right central scotoma; the left eye field is normal (lid artefact is visible (arrow)).
There was subsequent resolution of the right relative afferent pupillary defect but no improvement in his vision, and the question arose as to whether this was a traumatic retinopathy rather than an optic neuropathy.
Optical coherence tomography showed thinning of the outer nuclear layer of the retina (not shown).
Pattern electroretinogram, which reflects macular function (see box 3), was undetectable on the right and subnormal on the left. Full field electroretinograms were normal.
Retinal examination and imaging in the neuro-ophthalmology/eye clinic
Slit lamp examination
Examination with the slit lamp provides a high resolution, stereoscopic, inverted view of the disc and the fovea. Different lenses are used to optimise views of the peripheral retina.
Optical coherence tomography of the retina
Optical coherence tomography compares the scatter of low coherence infrared light waves reflected by the layers of the retina to a sample beam, allowing generation of high-resolution cross-sectional imaging of the retina. The relative thickness of each retinal layer can be mapped, and focal irregularities can be detected. It can be used as a diagnostic and a monitoring tool.
Autofluorescence
A metabolic mapping technique that uses light to detect metabolic by-products in the retina. Thus, hyperautofluorescence may indicate degenerative changes or oxidative stress and hypoautofluorescence may indicate cell loss.
Infrared imaging
Infrared light penetrates and is reflected differently in the retina in comparison with white light, enabling imaging of the outer retina. This technique makes abnormalities that are only subtly visible clinically or on colour photographs more apparent.
Angiography of the retina
Intravenous injection of an angiographic dye is used to delineate the normal vasculature of the retina and choroid, photographed using fluorescing light and captured through filters. Fluorescein dye is best for retinal vessels and indocyanine green is best for choroidal vessels due to differences in blood vessel walls. Dye leakage can indicate inflammation or neovascularisation, while hypofluorescence can be due to blockage of light (masking) or ischaemia in the affected area.
The electroretinogram
The electroretinogram is a diagnostic test that measures retinal electrical activity generated in response to a light stimulus. The basis for this is a retinal potential generated by light-induced changes in the flux of transretinal ions, primarily sodium and potassium. Electroretinograms are usually obtained using electrodes embedded in a corneal contact lens, which measure a summation of retinal electrical activity at the corneal surface. The technique can examine rod and cone function separately. Full field electroretinogram is useful for pan-retinal conditions but can miss focal pathology. Focal electroretinography is used to examine the macula, whereas multifocal electroretinography is used to examine the peripheral retina. A pattern electroretinogram has two components: the N95, which reflects ganglion cell function (similar to a retinal ganglion cell visual-evoked potential), and the P50, which reflects intermediate retinal layers. It can help to detect subtle optic neuropathies. (Usha R, updated 2015)16
Diagnosis: traumatic retinopathy
In this case, the initial relative afferent pupillary defect led to a diagnosis of optic neuropathy; however, its subsequent resolution resulted in further investigation, which demonstrated the retinopathy. There may have been an element of optic neuropathy initially. This case shows again the overlap in presentation between optic nerve and retinal conditions and at times there may be dual pathology.
Case 6
Part A
A 37-year-old myopic white woman, who had been previously well, experienced right-sided orbital pain that worsened with eye movements. Four weeks later, she developed blurred vision in the same eye. Her symptoms resolved spontaneously over a few weeks. Two months later, she developed similar symptoms in the left eye that again resolved over a few weeks. Over the next year, she experienced low-grade chronic ocular pain and redness of both eyes. Eighteen months after the initial presentation, her right eye became increasingly red and painful, and she complained of blurred vision with an intrusive yellow visual disturbance. Visual acuity was right eye 6/9 and left eye 6/4. Ishihara colour plates were 17/17 in the left eye but only 2/17 with the right eye. There was a subtle right relative afferent pupillary defect. On funduscopy there was optic disc swelling and macular oedema.
An MR scan of the brain and orbits with gadolinium, serum angiotensin-converting enzyme, antinuclear antibody, antineutrophil cytoplasmic antibody, antidouble stranded DNA antibody, anticardiolipin antibodies, rheumatoid factor, immunoglobulins, serum electrophoresis, lumbar puncture and gallium scan were normal.
The acuity and colour vision recovered to normal in 1 month. She was diagnosed with optic neuritis (third episode) with secondary macular oedema.
What are the atypical features for optic neuritis?
The most troubling is the normal enhanced MR scan. The optic disc swelling is relatively uncommon, but macular oedema is definitely atypical for demyelinating optic neuritis and can be seen in the various forms of atypical optic neuritis. As mentioned, mild optic disc swelling can occur as a secondary feature of ophthalmic conditions if the optic disc is involved in inflammation that is primarily intraocular (uveitis, scleritis or retinitis), and patients with macular or widespread retinal dysfunction may have a relative afferent pupillary defect, as may patients with optic neuropathy. Thus, this case has a wide differential diagnosis.
A retinal opinion was sought, for electrodiagnostic testing.
Part B
Slit lamp examination showed prominent deep scleral vessels in the right eye, indicating anterior scleritis. The anterior chambers and vitreous cavities were quiet. There were normal optic discs, and there were discrete areas of subtle retinal pigment epithelium atrophy at both posterior poles (figure 10A).
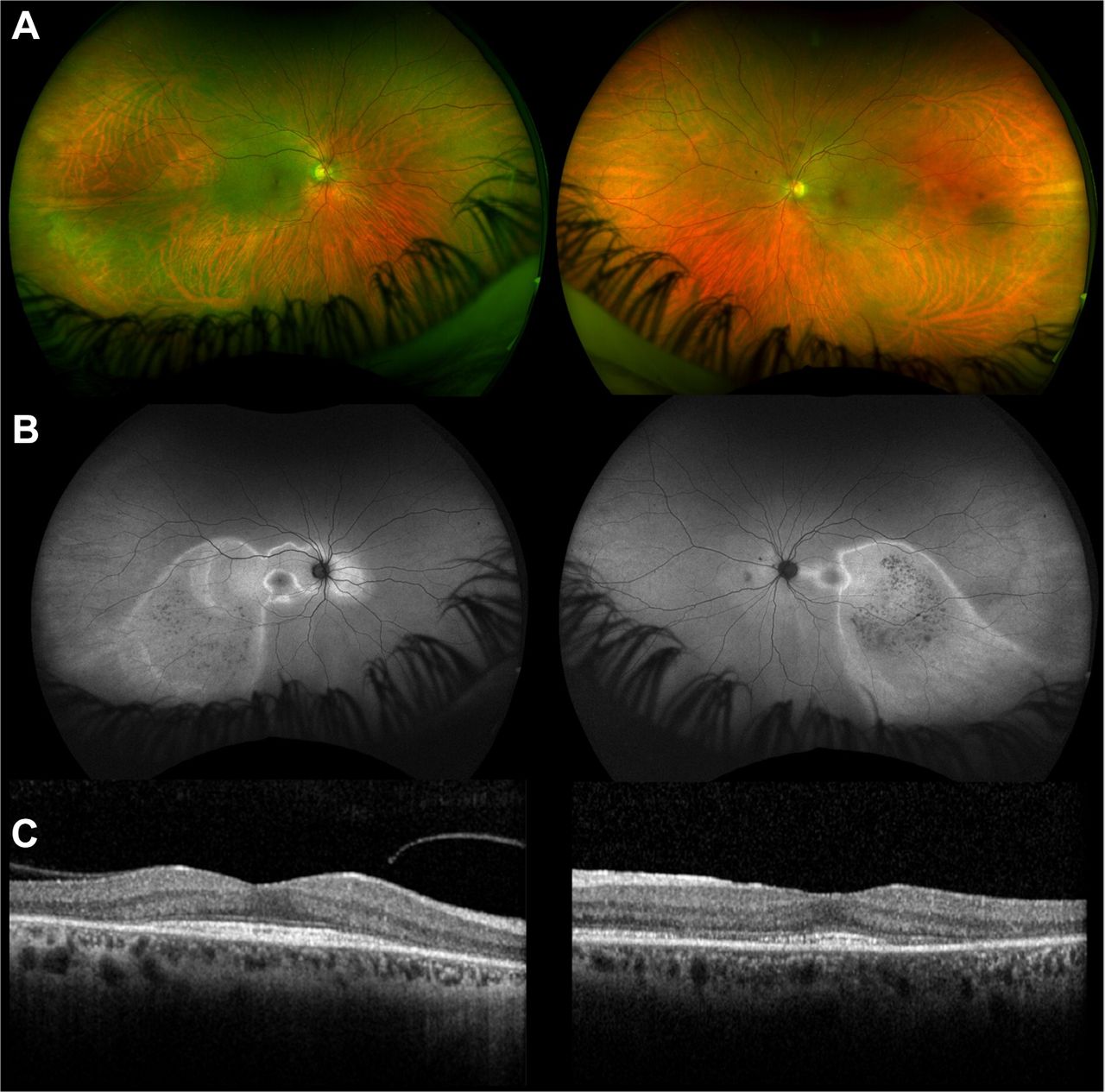
(case 6) (A) Fundus photography showing subtle discrete areas of RPE atrophy (green areas). (B) Autofluorescence imaging improves visualisation of the affected areas of retina, seen as zones of hyperautofluorescence. (C) Optical coherence tomography shows outer retinal (photoreceptor) disruption within the areas of affected retina (white arrows) with sparing at the foveal centre (yellow arrow). OCT, optical coherence tomography; RPE, retinal pigment epithelium
These changes were better seen using autofluorescence imaging with discrete areas of hyperautofluorescence corresponding with areas of outer retinal (photoreceptor) disruption on optical coherence tomography (figure 10B,C); there was greater foveal involvement in the left eye. An ultrasound scan of the right eye showed thickening of the posterior globe coats with fluid in sub-Tenon’s capsule space, indicating scleritis.
Visual evoked potentials were normal, but there was bilateral macular dysfunction shown on pattern electroretinogram. Multifocal electroretinography showed focal retinal dysfunction within the areas of affected retina, but full field electroretinograms were normal. Further blood tests, including syphilis serology and QuantiFERON-TB Gold, were normal. Antiretinal antibodies were not detected.
What are the most likely diagnoses?
Ocular pain and prominent deep scleral vessels suggest anterior scleritis. The ultrasound scan findings are diagnostic of posterior scleritis, which is commonly associated with secondary optic disc swelling. Posterior scleritis, in the absence of anterior scleritis, accounts for ~2% of all scleritis cases4 and can easily be mislabelled as optic neuritis.
However, the patient’s visual field defects and colourful photopsia, fundus imaging abnormalities and electroretinogram point to an additional retinopathy. The findings are consistent with a diagnosis of acute zonal occult outer retinopathy (AZOOR; see table 1).
We made a diagnosis of scleritis (having previously been misdiagnosed as optic neuritis) and AZOOR. We gave high dose oral prednisolone primarily to treat the scleritis; the ocular pain and redness rapidly resolved and the ultrasound scan normalised. Remission was maintained over a 7-year period on low dose prednisolone and occasional non-steroidal anti-inflammatory drugs.
Diagnosis: AZOOR, possibly triggered by scleritis
AZOOR usually occurs de novo, but a significant minority of cases are triggered by other pre-existing ocular inflammatory conditions (secondary AZOOR), such as punctate inner choroidopathy and multifocal choroiditis. There are no previously published cases of AZOOR associated with (or triggered by) scleritis.
Patients with AZOOR present with painless visual field defects and colourful photopsia, usually in the temporal quadrant, and blurred central vision if there is macular involvement.5 At presentation, 2/3 of cases are unilateral and funduscopy is normal in 90%; during follow-up, only 1/3 of all cases remain unilateral and 50% develop clinically visible zones of retinal pigment epithelium atrophy with pigment clumping.6
There are almost no high-quality therapeutic trials in AZOOR. Numerous therapies have been used, including immunosuppressive and antiviral agents but the results have been mostly disappointing. However, recently two groups independently reported a benefit from high dose systemic corticosteroids.7 8
Case 7
Part A
A 62-year-old man was referred by his general practitioner after his optician found a homonymous field defect at a routine appointment. He had a history of ‘retinal problems’ and dyslipidaemia and took atorvastatin.
He was referred to the ophthalmology clinic. Visual acuities were right eye 6/6 and left eye 6/9. Ishihara plate scores were right eye 12/13 and left eye 7/13. There was no relative afferent pupillary defect. Both optic nerves were within normal limits. Visual fields to confrontation showed a right superior quadrantic defect. There was evidence of bilateral maculopathy, likely old central serous retinopathy, but the changes were extensive and did not correspond to the visual field defect, which appeared homonymous. He was referred to neurology with a diagnosis of a postchiasmal lesion.
What is the diagnosis/diagnoses? What are the atypical features?
A sudden onset homonymous superior quadrantanopia in a man of this age suggests a lesion in the temporal portion of the optic radiation: the Flechsig-Meyer loop. The most common cause would be a lacunar infarct, but there is a broad differential diagnosis.
Although he is known to have a retinal disorder, one would also consider optic nerve pathology (reduced colour vision). However, the lack of relative afferent pupillary defect in particular raises the question of retinal pathology: a relative afferent pupillary defect can be masked by bilateral pathology but should be elicitable on careful examination when there is asymmetry.
MR scan of brain was normal. He was referred to the neuro-ophthalmology clinic.
Part B
Slit lamp examination both maculae showed significant pigment change.
Visual field defects were confirmed on automated perimetry, although there was a suggestion that the midline was not fully respected (see figure 11).

Humphrey field analyser (case 7). There is a homonymous field defect, although there is a suggestion that the defect in the right eye crosses the midline.
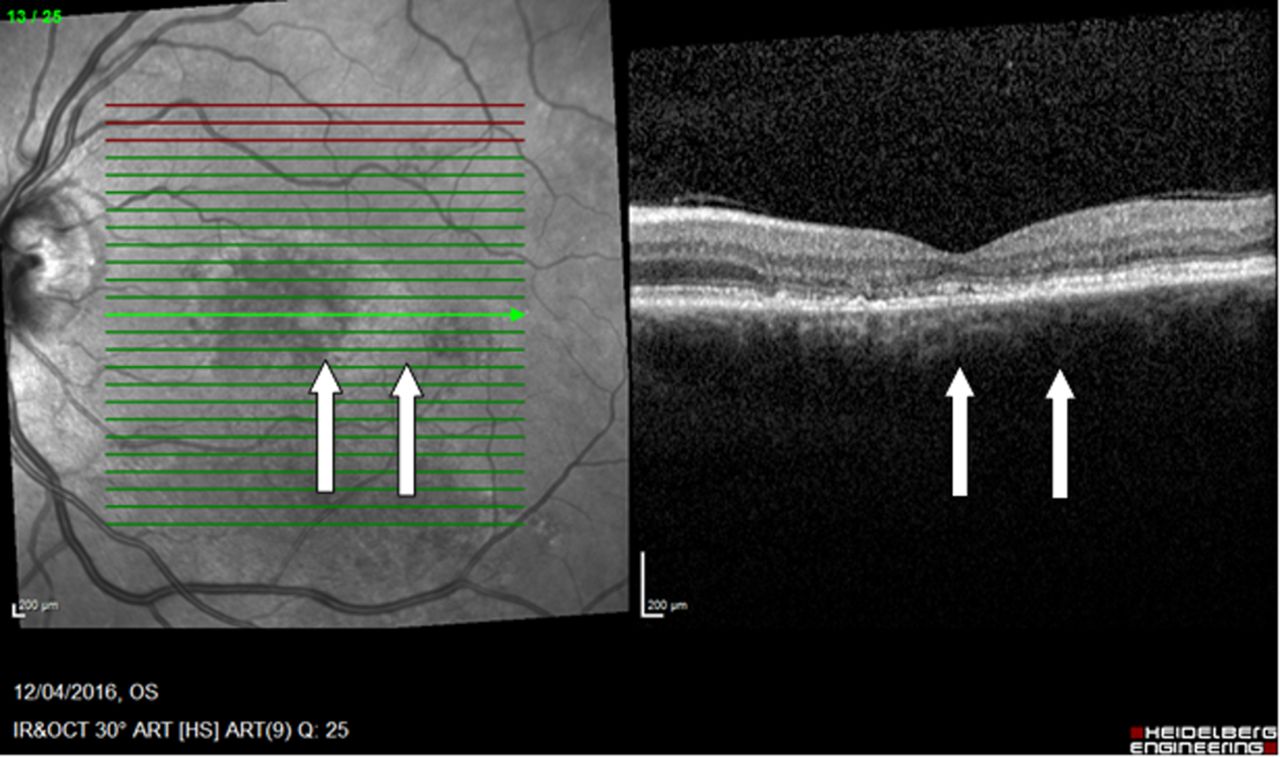
Fundus autofluorescence showed a pattern of outer retinal damage that was more extensive than the visual field loss (figure 12). However, macular optical coherence tomography showed obliteration of the photoreceptor layer close to the foveal centre in each eye (figure 13). This damage, which would result in dense visual field defects, precisely correlates with the pseudo-homonymous visual field loss (figure 11).

Autofluorescence left eye shown (case 7). The areas of hyperautofluorescence (demonstrating altered metabolism) are far more extensive than what is clinically apparent and demonstrated by the visual field defect in figure 10; this was the case bilaterally.

OCT left eye shown (case 7). On the cross-sectional OCT, which is centred on the macula, there is obliteration of the photoreceptor layer close to the fovea (arrows). This region extended inferiorly in a vertical strip temporal to the fovea (superonasal visual field). In the right eye, there was a corresponding strip nasal to the fovea (superotemporal visual field). These portions of the overall pathology in each eye correspond precisely to the pseudo-homonymous visual field defect shown in figure 11. OCT, optical coherence tomography.
It later came to light from the patient’s records that he had indeed a history of central serous chorioretinopathy that had affected both eyes.
Diagnosis: central serous retinopathy causing a pseudo-homonymous haemianopic scotoma and dyschromatopsia
Central serous chorioretinopathy is a type of acute maculopathy caused by accumulation of subretinal fluid at the macula. It typically affects men aged 20–50 years and is associated with corticosteroid exposure and ‘type A’ personality traits. Patients can present with central scotoma, metamorphopsia and reduced colour vision. On examination, there is characteristic serous detachment of the neurosensory retina caused by fluid leakage from the choriocapillaris through the retinal pigment epithelial layer. Other causes for retinal pigment epithelial leaks, such as choroidal neovascularisation, should be ruled out. Demonstration of distortion is often critical in clinically differentiating central serous chorioretinopathy from optic neuritis.
Other acute maculopathies associated with retinal pigment epithelial layer leak
The most common maculopathies in the UK are age-related macular degeneration and diabetic macular oedema, in which patients may present acutely with reduced vision, central scotoma and distortion, usually with a background of pre-existing age-related macular degeneration or poorly controlled diabetes mellitus.
Other acquired maculopathies that can be associated with acute visual loss include:
Solar retinopathy, in which central visual loss occurs within 4 hours of staring at the sun, with a small yellow spot often visible at the fovea.
Valsalva retinopathy, in which retinal haemorrhages develop after severe straining.
Idiopathic polypoidal choroidal vasculopathy, which is similar to age-related macular degeneration, except it is often unilateral and occurs in Asians rather than Caucasians.
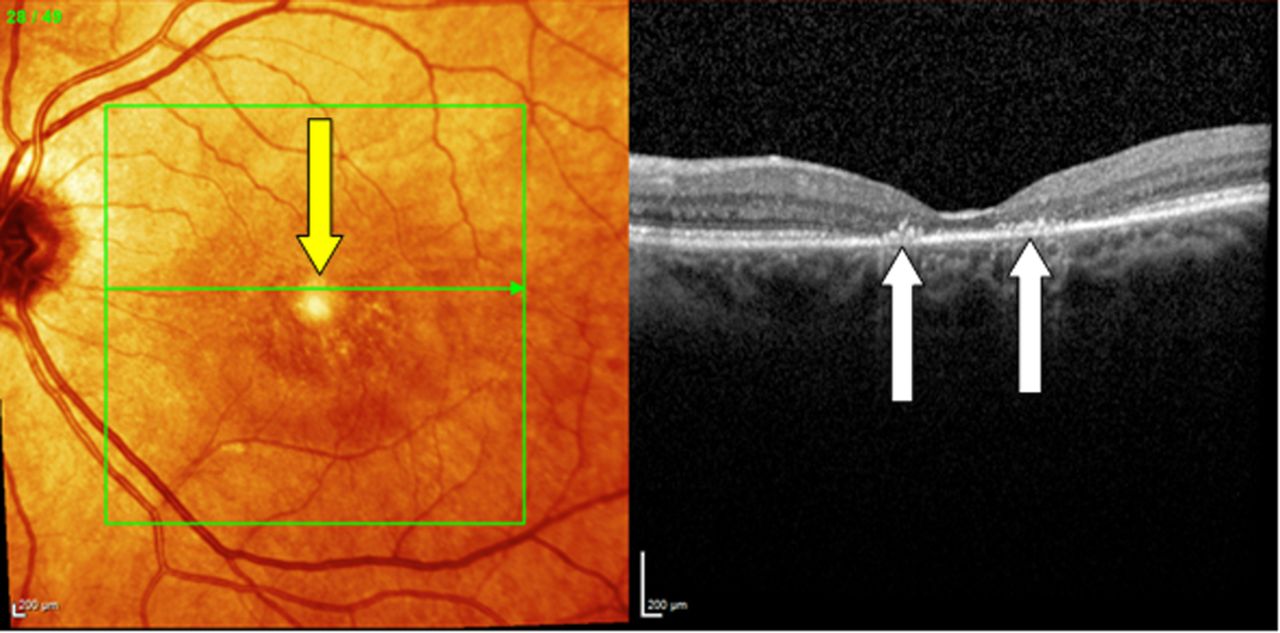
‘Poppers retinopathy’, which presents with sudden onset bilateral central visual loss due to a toxic effect of alkyl nitrite compounds on central photoreceptors, usually in young males. Poppers remain popular in the UK among clubgoers.9 Fundus changes are subtle, but the optical coherence tomography appearance is diagnostic (see figure 14).

‘Poppers’ retinopathy. The cross-sectional OCT, centred on the macula, demonstrates clear disruption of the inner/outer retinal segment. The patient presented with sudden bilateral visual loss after inhalation of ‘poppers’, likely isopropyl nitrite, which is toxic to central photoreceptors and was referred to neurology from an eye casualty as bilateral optic neuritis. OCT, optical coherence tomography.
Case 8
Part A
A 25-year-old man presented to eye casualty with a 4-week history of bilateral blurred vision. There was no associated pain, redness or photophobia, but he described a flickering light in his central vision. He was referred to neurology as a possible bilateral optic neuropathy.
He had no headaches or neurological symptoms, and there was no associated viral illness. He had been previously well and took no regular medications.
On examination, he had visual acuities of 6/9 in both eyes, 17/17 Ishihara plates were read bilaterally and there was no relative afferent pupillary defect. Visual fields to confrontation with a red hat pin revealed small paracentral scotomata. Funduscopy showed normal optic disc appearance and yellow-white lesions at the posterior pole in both eyes. His neurological examination was normal.
Where is the lesion?
The scotomata are less likely to be due to a lesion in the optic nerve in the absence of a relative afferent pupillary defect, but this will only be detectable in bilateral optic neuropathy if the visual loss is sufficiently asymmetric. The positive symptom and visible retinal abnormality suggests that the central scotomata arise from a maculopathy.
Part B
On slit lamp examination, there were bilateral yellow-white placoid lesions in the macula area but no other signs of intraocular inflammation. The lesions were seen more clearly in infrared imaging (see figure 15).
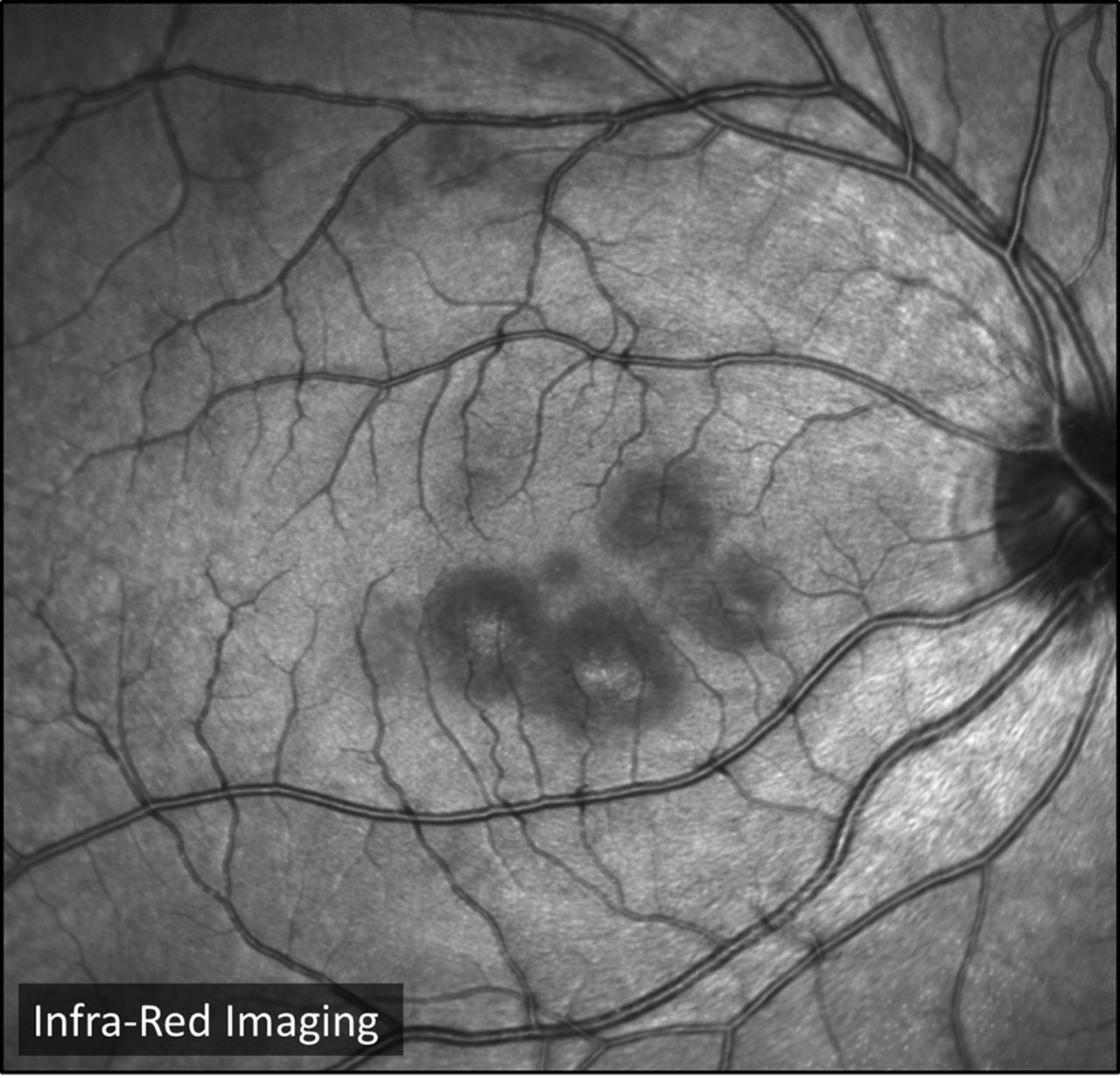
Infrared image of the right eye (case 8). Infrared images are taken using a confocal scanning laser ophthalmoscope. The lesions are visible as darker patches surrounding the fovea, which also appears dark.
Ocular coherence tomography showed outer retinal changes in the same area as the visible lesions (see figure 16). Figure 17 shows the angiographic findings.
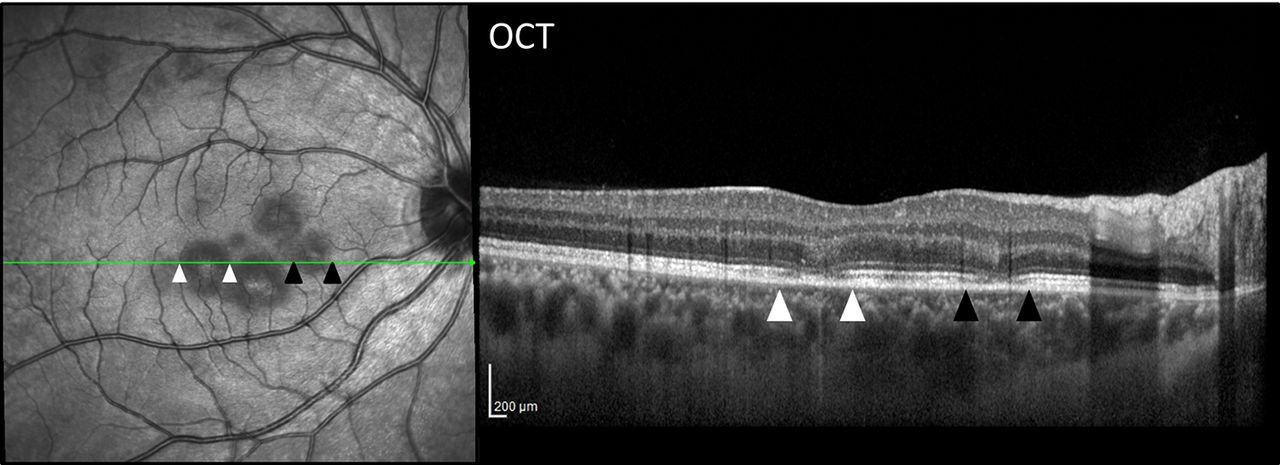
OCT of the right eye (case 8). The cross-sectional OCT centred on the macula demonstrates disruption of the outer retinal layers between the white and black arrowheads. These are seen to correspond to the funduscopically visible lesions. OCT, optical coherence tomography.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Indocyanine green angiography (ICG) and fluorescein angiography (FA) of the right eye (case 8). FA and ICG on the same day. Multiple dark lesions are seen on the ICG. FA demonstrates initial hypofluorescence then in later phases hyperfluorescence over the lesions; here the late phase FA shows staining and leakage of dye over only the largest lesions. The red arrows highlight a new choroidal lesion on ICG, which has produced no visible retinal changes on the FA.
What is the differential diagnosis?
White dot syndromes (see table 1).
Acute macular neuroretinopathy—typically young or middle-aged women with the acute onset of scotomata that may be bilateral. Multiple characteristic dark, well-defined, wedge-shaped intraretinal lesions pointing to the fovea, often in a flower petal arrangement, sparing the retinal pigment epithelial layer.
Infective: syphilitic placoid chorioretinitis or tuberculous choroiditis.
Blood tests for syphilis, tuberculosis and an autoimmune screening panel were negative. After 1 month, his visual acuity had improved to 6/6 in both eyes; after 3 months, he was asymptomatic, and the lesions had fully resolved. We diagnosed acute posterior multifocal placoid pigment epitheliopathy (APMPPE), based on the characteristic imaging findings and the exclusion of the differential diagnoses.
Diagnosis: APMPPE
APMPPE is an idiopathic retinopathy that typically affects young adults aged 20–50 years, has an equal sex distribution and in one-third is preceded by a viral illness. The symptoms develop rapidly; if the lesions involve the fovea, these can include marked visual acuity loss or paracentral scotomata and photopsia. Two-thirds of cases have a bilateral presentation; in most cases the lesions resolve spontaneously, and visual acuity returns to normal over about a month.10 Increasingly, patients with foveal lesions and profound visual loss receive oral corticosteroids, which may hasten recovery, although there are few scientific data to confirm this.
Clinical features include the typical funduscopy findings of multifocal large creamy white placoid lesions in the posterior pole. The visible lesions appear at the level of the retinal pigment epithelial layer with optical coherence tomography showing disruption of the outer retinal layers (figure 16). Fluorescein angiography shows initial hypofluorescence; in later phases, there is hyperfluorescence over the lesions. Figure 17 shows these changes in comparison with an indocyanine green angiogram on the same day. The red arrow highlights one of many lesions visible as dark hypofluorescence on this angiogram, not present in the fluorescein angiogram. Because indocyanine green angiography highlights choroidal pathology—which occurs earlier and more extensively—the prevailing theory is that the initial acute inflammatory response occurs in the choriocapillaris.11
A delayed-type hypersensitivity reaction as the pathophysiology would also explain the associations described with other vasculitic conditions including cerebral vasculitis.12 Every patient with APMPPE should be questioned about headaches, limb weakness or paraesthesia and may need appropriate neurological investigations.
Conclusions
Retinal problems may be referred to neurologists as possible neurological conditions such as optic neuritis. Symptoms and clinical findings can overlap. However, with careful history taking and examination, the pathology can be localised and sometimes diagnosed in the general neurology clinic without slit lamp examination. Timely recognition of these patients and referral to a retinal specialist is important for optimal management.
White dot syndromes
This group of conditions is defined by chorioretinitis of the outer photoreceptor layer, often in association with an enlarged blind spot. There is some debate as to whether these are distinct conditions or overlapping syndromes that should all come under the umbrella of acute zonal occult outer retinopathy (table 1).17
References
Footnotes
Contributors ZK: acquisition and collation of cases and photographs and drafting and revision of manuscript. WRT, LdP and JDV: Acquisition of case and photographs, drafting and revision of manuscript. GTP: conception and design of article, acquisition and selection of cases and revision of manuscript.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed by Mark Lawden, Leicester, UK.
Other content recommended for you
- Mimics and chameleons of optic neuritis
- Placoid lesions of the retina: progress in multimodal imaging and clinical perspective
- Simultaneous presentation of multifocal choroiditis and acute zonal occult outer retinopathy in one eye
- Persistent photopsia: multiple evanescent white dot syndrome in a sexagenarian
- Retinal imaging: what the neurologist needs to know
- Multiple evanescent white dot syndrome: clinical course and factors influencing visual acuity recovery
- Correlation of spectral optical coherence tomography with fluorescein and indocyanine green angiography in multiple evanescent white dot syndrome
- Acute zonal occult outer retinopathy: towards a set of diagnostic criteria
- Multimodal imaging findings in ‘hyper-early’ stage MEWDS
- Choroidal neovascularisation triggered multiple evanescent white dot syndrome (MEWDS) in predisposed eyes