Article Text

Abstract
We describe a case of functional vitamin B12 deficiency where the repeated measurement of a serum B12 level within the normal range led to delay in the diagnosis of subacute combined degeneration of the spinal cord, and possibly permanent neurological damage as a result. Failure of intracellular transport of B12 by transcobalamin-2 can lead to functional B12 deficiency but with apparently normal serum levels, and is suggested by raised levels of either serum methylmalonic acid or homocysteine, associated with low levels of transcobalamin-2. Such patients may respond to repeated high-dose injections of B12.
Statistics from Altmetric.com
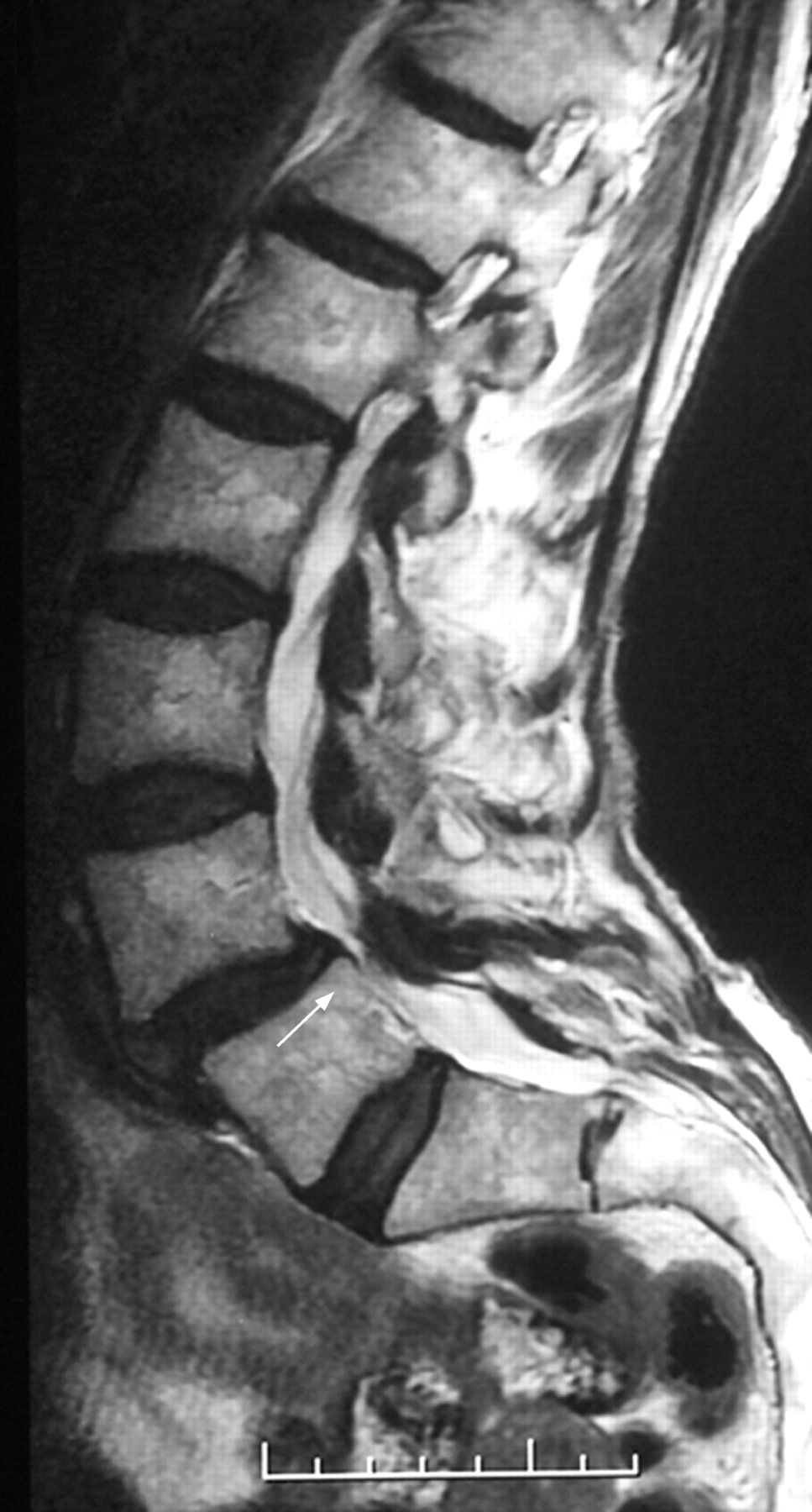
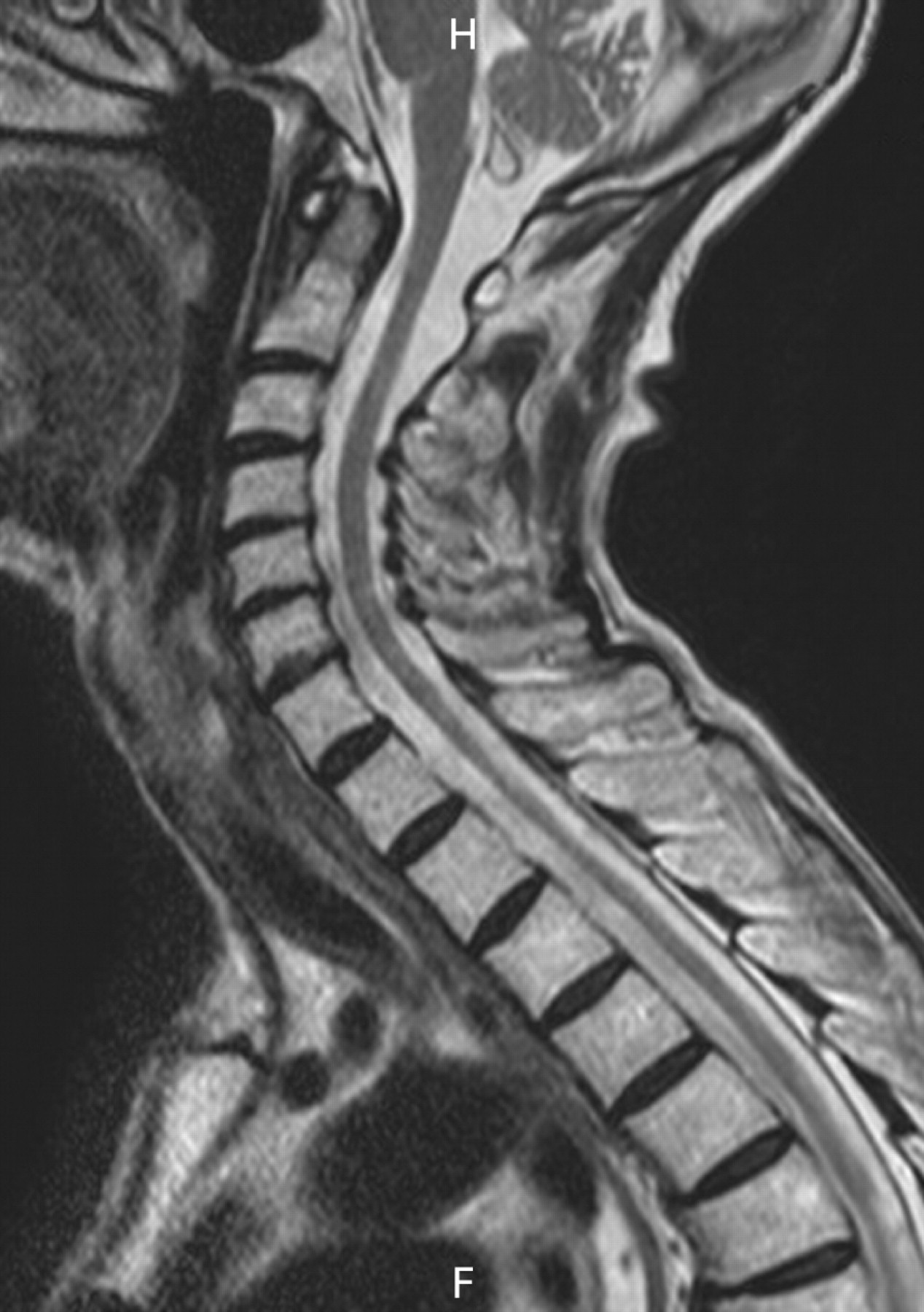
A 73-year-old woman was admitted to hospital following a fall in dim lighting conditions. She gave a 10-year history of progressive gait deterioration associated with urinary urgency. Examination revealed a spastic paraparesis with marked distal proprioceptive loss. The upper limb and cranial nerve examination were normal. Her past medical history included a right mastectomy and local radiotherapy for breast carcinoma 16 years previously, and a myocardial infarction 10 years prior to this admission. There was no family history of neurological disease. She had been referred for a neurological opinion three years previously, reporting symptoms suggestive of lumbar spinal claudication in the form of progressive leg weakness after walking 100 yards, relieved by 30 minutes of rest. MRI of the entire spine at that time revealed significant lumbar canal stenosis with notable disc protrusion at L5 (fig 1). The patient underwent an uncomplicated laminectomy, but her gait continued to deteriorate.
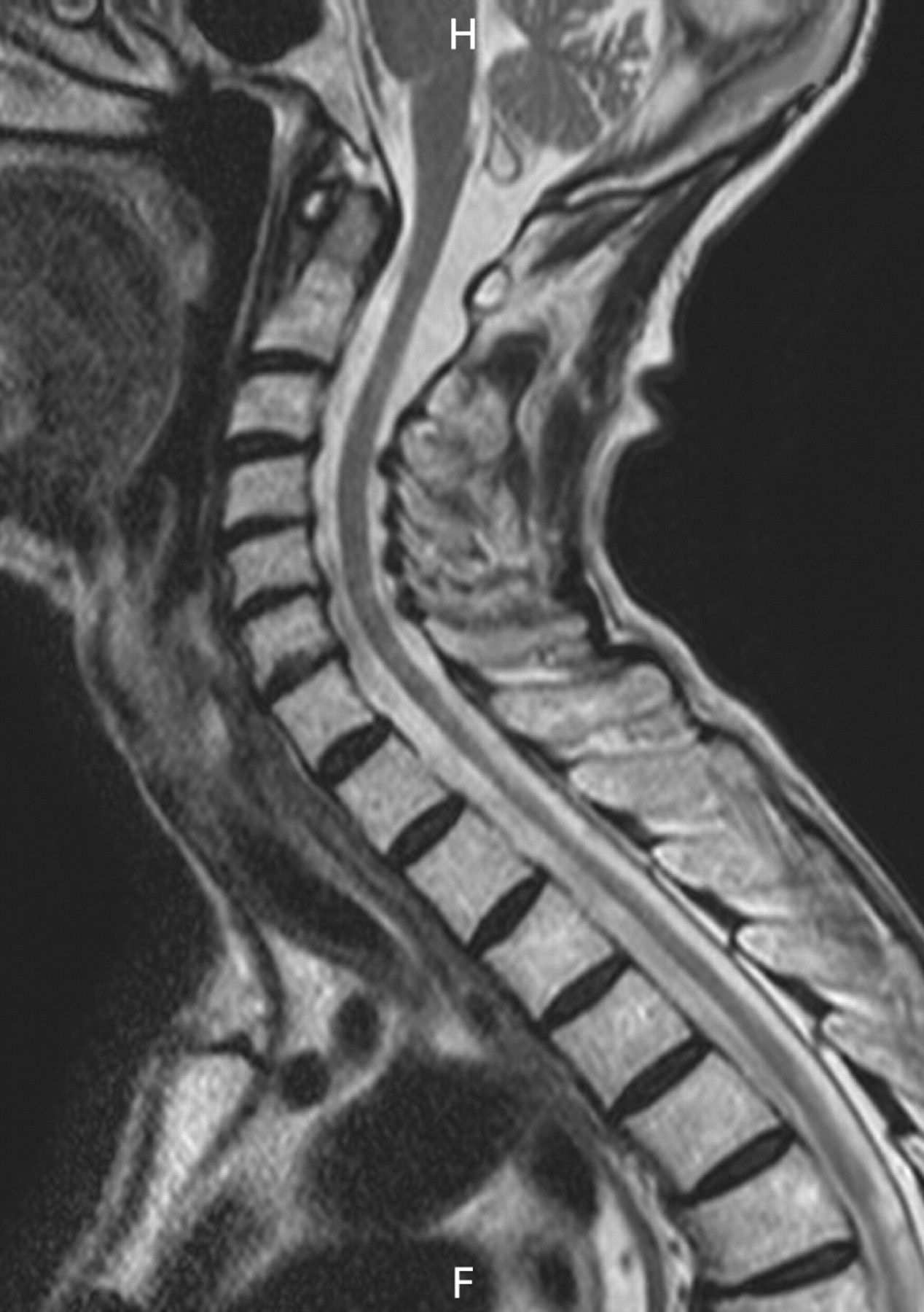
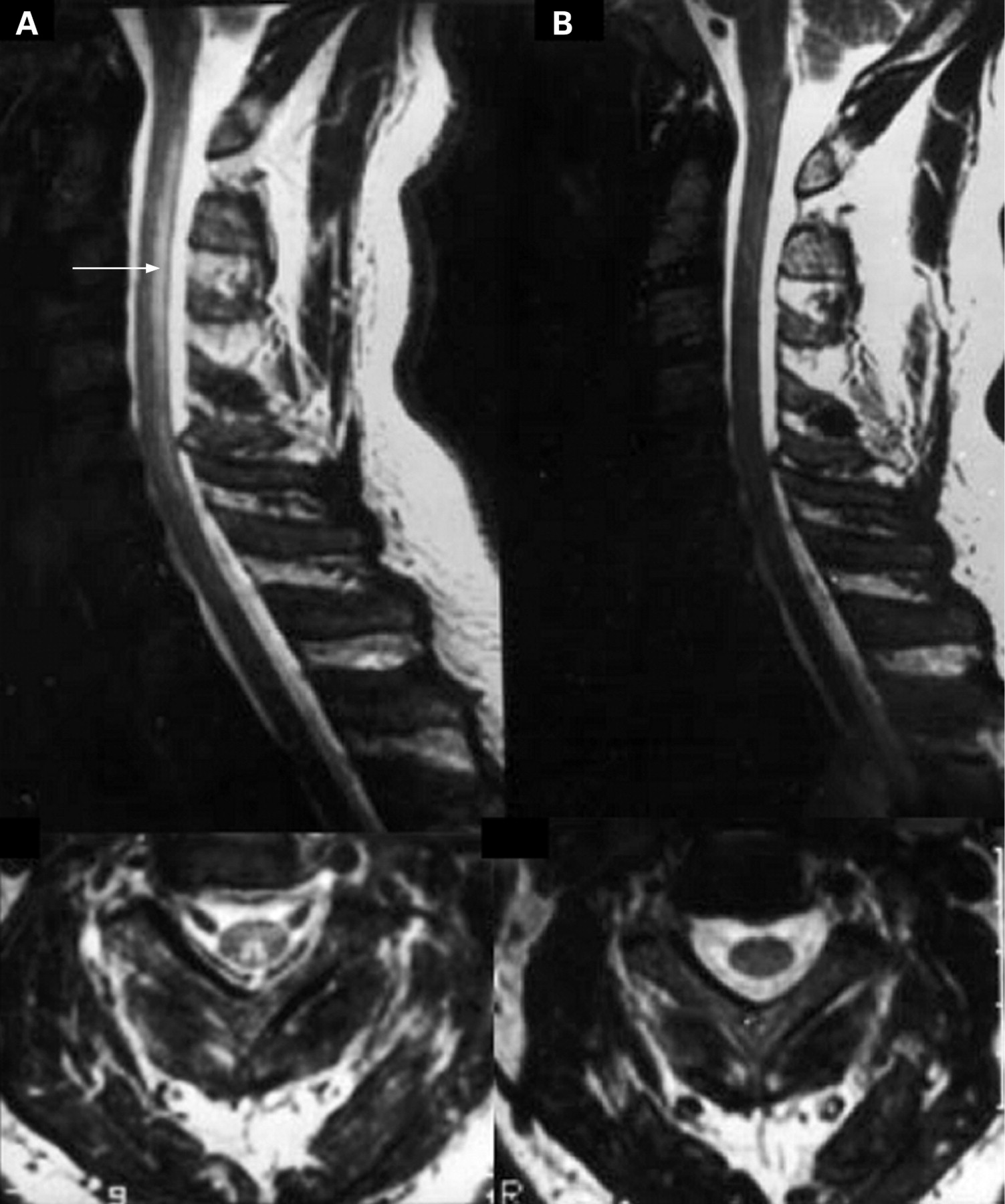
Repeat MRI of the entire spine and a CT of the brain on this admission were unremarkable. No marked high intensity signal change was seen in the cervical cord (fig 2).
Serum vitamin B12 measured at the time of her initial presentation three years before was 349 ng/l (normal range 180–900). A repeat level during this latest admission was also within the normal range at 250 ng/l. The haemoglobin was 8.7 g/dl and mean cell volume 98. Serum folate levels were normal on both occasions. The serum copper level was normal at 20 μmol/l (normal range 11–20). However, the serum methylmalonic acid (MMA) level during the latest admission was markedly raised at 722 nmol/l (normal range 73–271), suggesting functional vitamin B12 deficiency.
The patient was started on three-monthly intramuscular injections of 1 mg vitamin B12 (hydroxycobalamin), after initial alternate day loading. After six months there had been little functional improvement but no further decline either. A repeat MMA level was markedly lower, though still slightly raised at 316 nmol/l, with a normal homocysteine level of 12.7 μmol/l (normal range 5–15), suggesting improved tissue availability of vitamin B12. Transcobalamin (TC) levels were measured shortly after starting treatment. TC-1 was slightly raised at 149 ng/l (49–132), TC-2 markedly reduced at 202 ng/l (402–930), TC-3 normal at 117 ng/l (80–280) and overall TC unsaturated binding capacity reduced at 446 ng/l (520–1132)—all felt to be consistent with functional B12 deficiency.
DISCUSSION
This patient presented with clinical features consistent with subacute combined degeneration of the cord, namely a slowly progressive spastic paraparesis with distal proprioceptive loss, without a compressive or inflammatory cause on MRI or any family history of hereditary spastic paraparesis. In this case the clinical evolution was more drawn out than would normally be accepted under the term “subacute”. Nonetheless, the median time to diagnosis from symptom onset in neurological presentations of B12 deficiency in general is approximately one year, with a range extending to more than four years.1 Although the diagnosis and treatment of lumbar canal stenosis was supported by the recorded clinical and MRI findings, in retrospect we suspect this may have been a secondary diagnosis in addition to established subacute combined degeneration, given the continued deterioration post-laminectomy. Post-radiotherapy late effects were considered as a cause, but the cord was not within the field of the beam tattoo markings, and there were no features of radiation myelopathy on imaging.
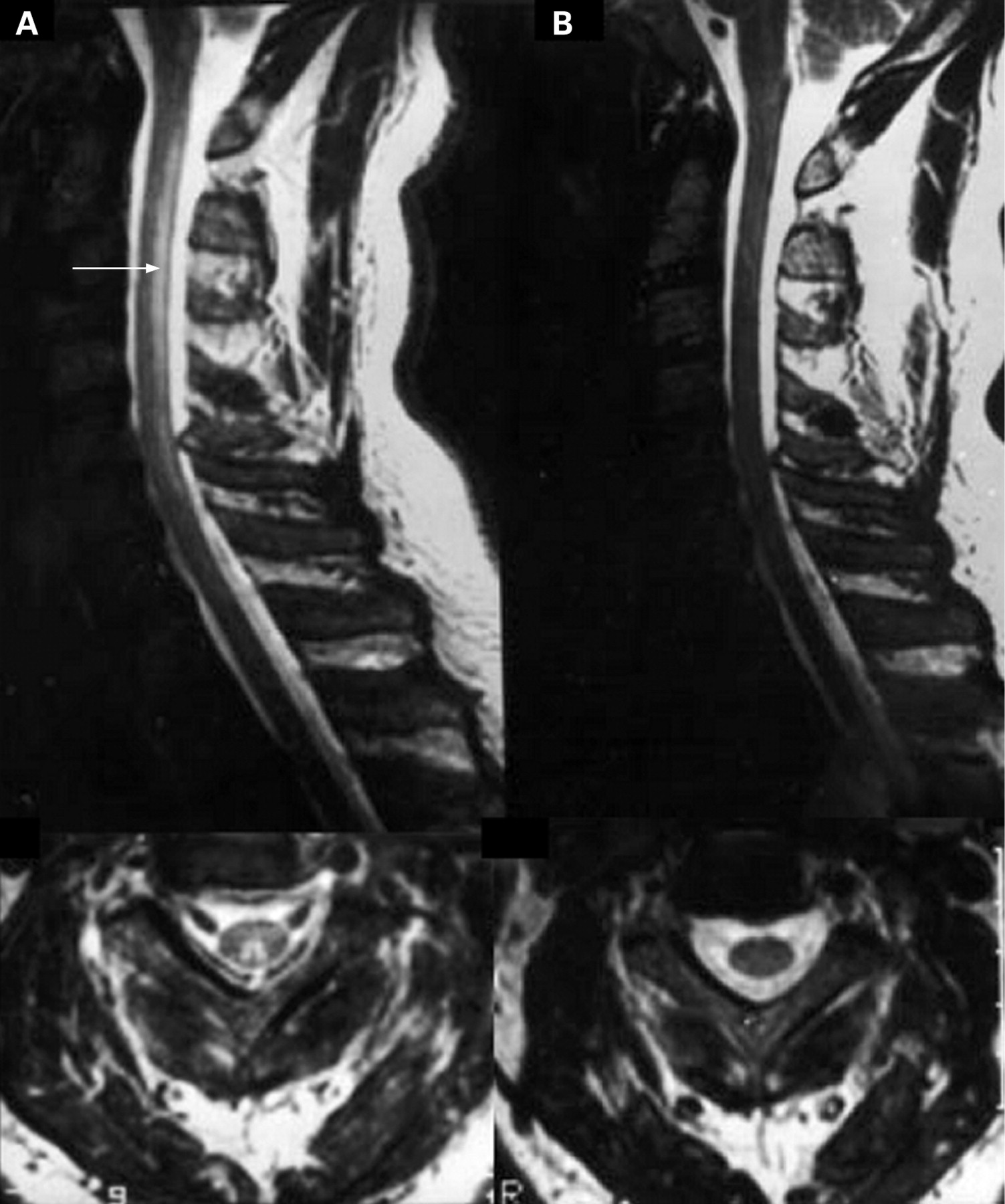
Subacute combined degeneration of the cord is probably best considered a clinical syndrome with a number of causes, although the differential diagnosis is very limited, vitamin B12 deficiency accounting for most cases, with copper deficiency and tertiary syphilis much rarer causes. Hyperintense signal change may be seen within the dorsal columns of the cord in cases of subacute combined of any cause (fig 3), and a variety of peripheral as well as central neurophysiological abnormalities have been described in more typical B12 deficiency.12
Vitamin B12 deficiency is most often detected by serum B12 measurement alone, and such deficiency may be associated with auto-antibodies to either gastric parietal cells or the intrinsic factor required for vitamin B12 binding. The term “pernicious anaemia” specifically refers to B12-related megaloblastic anaemia in the setting of atrophic gastritis. As well as autoimmune, B12 deficiency may be dietary, malabsorptive due to bacterial overgrowth and any cause of villous atrophy; or Scandinavian fish tapeworm-related. George Whipple (fig 4), George Minot and William Murphy shared the 1934 Nobel Prize for Medicine for their discovery of liver (containing high levels of vitamin B12) as a treatment for pernicious anaemia. Alexander Graham Bell was a famous sufferer (fig 5).


An important point is that the absence of anaemia with or without macrocytosis does not necessarily exclude the diagnosis of B12 deficiency. In this case the anaemia was attributed to acute blood loss from a significant scalp laceration sustained in the fall of the second presentation, and the haemoglobin level returned to normal within four months. Serum vitamin B12 levels were well within the normal range at both presentations in our patient, with no evidence of megaloblastic anaemia.
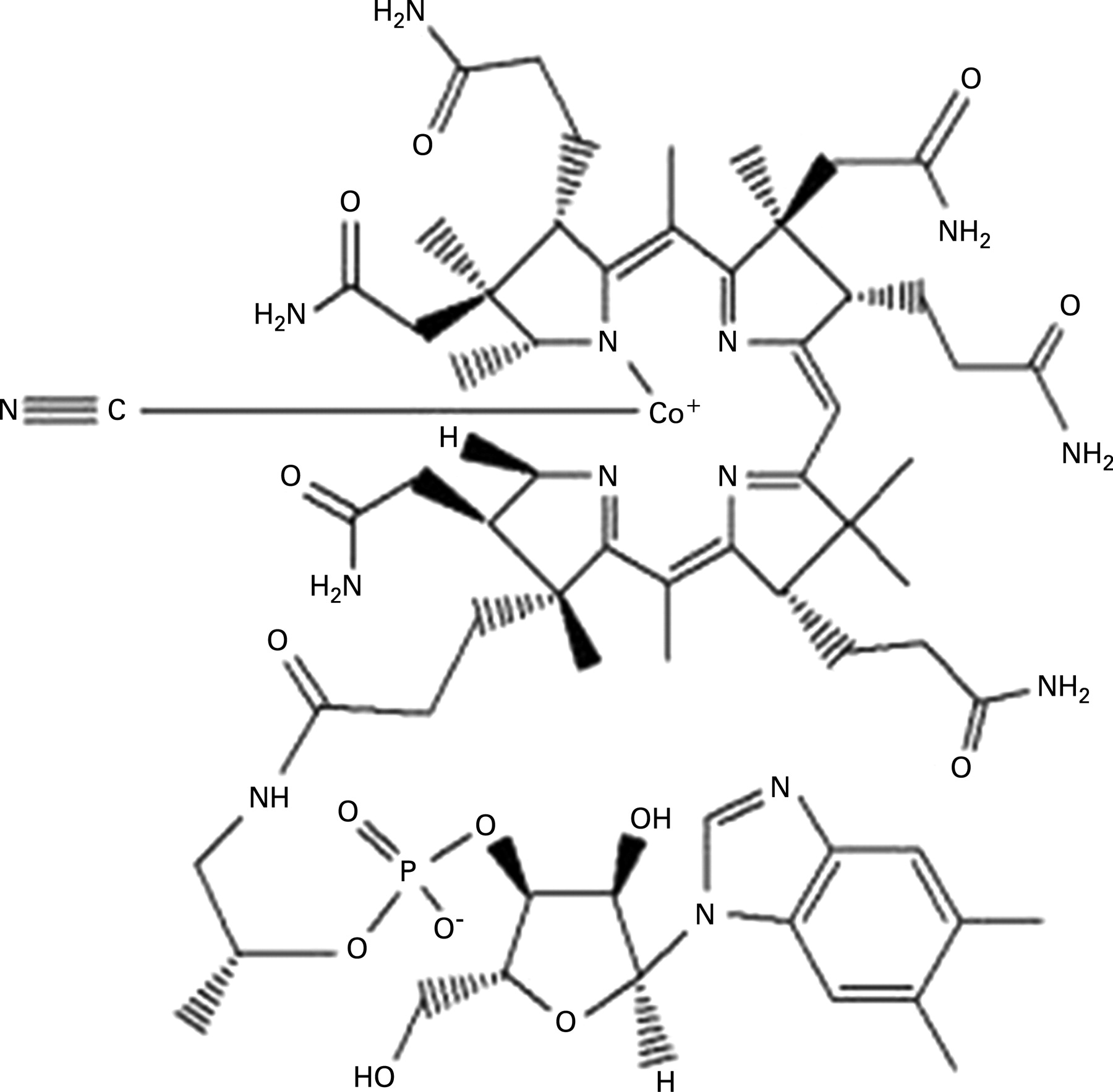
Vitamin B12 encompasses several compounds, termed cobalamins (fig 6), and these have an intense red colouration. In combination with folic acid, cobalamins are essential co-factors for DNA synthesis, notably within the bone marrow and myeloid cells, but also critical to the process of neuronal myelination. Cobalamins facilitate mitochondrial methylmalonylcoenzyme A mutase conversion of MMA to succinate, linking carbohydrate and lipid metabolism; and also the activation of methionine synthase in the conversion of homocysteine and methyltetrahydrofolate to methionine. Raised levels of MMA and/or homocysteine therefore may reflect a functional deficiency of cobalamins, although caution must be used in the presence of significant renal impairment (our patient had a normal predicted glomerular filtration rate).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cobalamins are plasma-bound, largely to transcobalamin (TC)-I and -2, and the latter promotes cellular uptake by TC receptor-mediated endocytosis. A common TC-2 gene 776C>G polymorphism, for which 41% of white people are heterozygous, is postulated to influence vitamin B12 tissue delivery and functional status, with heterozygotes being significantly impaired in this respect. Complete TC-2 deficiency is an autosomal recessive condition characterised by normal serum B12 levels but severe infantile megaloblastic anaemia and neurological damage.3 The TC genotype was not established in our case, but there was no apparent family history, and the low level of TC-2 and TC binding capacity provided strong indirect evidence for functional deficiency via this mechanism.
Although there is currently no simple monogenetic model for functional deficiency of vitamin B12, there is justifiable concern that serum vitamin B12 levels are a poor marker of the target tissue availability of cobalamins, so measuring serum MMA or homocysteine levels is a more accurate reflection of functional status. The markedly raised levels of MMA in our patient, and the clear biochemical response to exogenous hydroxycobalamin administration, support a diagnosis of functional cobalamin deficiency. The lack of clinical improvement after six months may reflect a very slow path to recovery, or the fact that chronic damage over many years is now permanent. At present, we continue to be encouraged by the lack of deterioration.
We were unable to identify any similar published cases. Gale and colleagues reported a 72-year-old patient presenting with confusion, clinical subacute combined degeneration of the cord and megaloblastic anaemia.4 Normal serum B12 levels were found despite later detection of intrinsic factor antibodies in the serum. A markedly elevated serum homocysteine raised the suspicion of an additional functional deficiency, and she was found to be heterozygous for the 776C>G polymorphism of the TC-2 gene. She received exogenous intramuscular B12 injections without initial improvement, but then underwent an apparent “Lazarus-like recovery” after the administration of corticosteroids. The authors attributed the latter to an effect on reducing the intrinsic factor antibodies postulated to be of such high affinity to crossreact with the circulating TC-2, antagonising tissue cobalamin uptake as a result.
Practice points
A normal serum vitamin B12 level does not accurately reflect the underlying tissue availability of cobalamin and may be falsely reassuring.
In the presence of a clinical picture suggestive of subacute combined degeneration of the spinal cord, methylmalonic acid or homocysteine levels should be measured to exclude functional vitamin B12 deficiency.
EDITOR’S NOTE
Occasionally I think it is very illuminating to publish a reviewer’s comments when there is an interesting area of disagreement with the authors of a paper, and here they are: “With transcobalamin II levels reduced to 50% of normal, and a raised serum methylmalonic acid concentration, partially reversible with vitamin B12, this patient has evidence of functional vitamin B12 deficiency. This may well have been caused by a heterozygous transcobalamin II gene polymorphism, though the patient’s genotype is not given. However, there remains concern about the extent to which this functional deficiency contributed to the patient’s neurological presentation. Several clinical features would be atypical for B12 deficiency—chronicity of the symptoms, early sphincter involvement, sparing of the upper limbs, and lack of positive sensory symptoms. These findings raise the question of dual pathology. Among other possibilities, late-onset primary progressive multiple sclerosis enters the differential diagnosis. There are no comments on MRI brain findings (admittedly potentially difficult to interpret in this patient’s age group) or a CSF examination.”
So maybe there was another pathology lurking somewhere, and not just functional B12 deficiency. Sorting out obscure myelopathies is not easy, and not as far as I know of very much research interest. But there is a good article in Practical Neurology to refer to: Wong SH, Boggild M, Enevoldson TP, et al. Myelopathy but normal MRI: where next? Pract Neurol 2008;8:90–102.
Acknowledgments
This article was reviewed by Lionel Ginsberg, London, UK.
Other content recommended for you
- Whippits, nitrous oxide and the dangers of legal highs
- Nutritional peripheral neuropathies
- Homocystinuria diagnosis and management: it is not all classical
- Progressive unsteadiness in a 68-year-old man with longstanding abdominal pain and altered bowel habit
- Vitamin B12 and folate deficiency in chronic heart failure
- Vitamin B12 deficiency
- Diagnosis of Vitamin B12 Deficiency in Patients With Myeloproliferative Disorders
- Important reminder: nitrous oxide (N2O) as a cause of functional vitamin B12 deficiency
- The difficulties with vitamin B12
- Nitrous oxide myelopathy with functional vitamin B12deficiency