Article Text

Abstract
Sporadic Creutzfeldt–Jacob disease (CJD) is a rare untreatable neurodegenerative disease which every neurologist will occasionally encounter during their career. However, it is likely to appear on their differential diagnosis list significantly more frequently. Numerous conditions can present with subacute encephalopathy which might be sporadic CJD and this article explores these diagnoses. It includes the commonest sporadic CJD mimics which are neurodegenerative, and highlights the relatively rare treatable mimics which must not be missed. It discusses relevant investigations, including serum antibodies, CSF, electroencephalography and MR brain imaging, and strategies when preliminary investigations fail to support sporadic CJD but no alternative diagnosis is readily apparent.
Statistics from Altmetric.com
Introduction
Sporadic Creutzfeldt–Jacob disease (sCJD) is a neurodegenerative, uniformly fatal prion disease characterised pathologically by spongiform change within the brain. The cause is unknown. It is rare, with an annual incidence in the UK of 1–1.5/million, meaning that on average an individual UK neurologist may encounter a new case only once every 5 years. However, far more patients present with a subacute encephalopathy which might be CJD. Here I will discuss such patients and their differential diagnoses, particularly those disorders which are amenable to treatment.
Most infective encephalopathies are too acute to be confused with CJD
The diagnostic possibilities
There is a wide differential for subacute encephalopathy (table 1). However, a large number of potential causes can be confirmed or excluded fairly quickly with a detailed history, including of drug and alcohol use, and some simple investigations such as routine bloods, CSF and CT brain imaging (eg, drug/toxic encephalopathy, brain metastases, systemic and common CNS infections and many metabolic causes). Most infective encephalopathies are too acute to be confused with CJD but rarely even common causes of encephalitis, such as herpes simplex virus (HSV) 1 and 2, can present as a subacute encephalopathy. Clearly, if there is concern about herpes simplex encephalitis it is prudent to treat with acyclovir until the diagnosis is excluded (usually by a negative HSV PCR on CSF). However, the clinical picture of fever, headache, reduced consciousness with CSF pleocytosis in addition to sometimes focal changes on MRI usually makes the diagnosis of an infective process relatively straightforward. The situation becomes more complicated in the immunocompromised patient or returning travellers, with a much wider differential diagnosis which is beyond the scope of this article.1
Differential diagnosis of subacute encephalopathy
Sporadic CJD
The mean age of sCJD disease onset is 66 years with most cases presenting between the ages of 50 and 75 years although patients as young as 142 and as old as 863 have been reported. There is no gender difference. Disease duration is typically short, with a median of 6 months from onset to death. Only 14% of cases survive longer than a year and only 5% live for 2 years or more.4
Clinical picture
The characteristic clinical picture in sCJD is one of rapidly progressive dementia with associated neurological features, particularly cerebellar ataxia, pyramidal signs and myoclonus. Visual disturbance, ocular movement disorders, extrapyramidal signs and hallucinations are also well recognised. The final stages are characterised by an akinetic mute state. The illness may be preceded by a non-specific prodrome, including fatigue, low mood, weight loss and headache for a few months. Although the commonest sCJD presentation is with a subacute encephalopathy, other onsets include pure cerebellar, visual, psychiatric and stroke-like syndromes. The rapidity of deterioration in sCJD usually helps distinguish it from other dementias and neurodegenerative conditions.
Investigations
There are no specific findings on blood tests; their primary value is to exclude other diagnoses.
The CSF is usually unremarkable with normal glucose and no cells although protein may be modestly raised (usually less than 1 g/l).5 A raised white cell count virtually excludes sCJD. The most valuable CSF test is analysis of the neuronal protein 14-3-3 which has a sensitivity of 90–97% and specificity of 87–100% in patients with suspected sCJD referred to a CJD surveillance unit.6 7 Confirmed sCJD cases with a normal 14-3-3 are often clinically and pathologically atypical, with a younger age of onset and prolonged disease duration. The value of the test is much less when used as a screening tool for unselected patients with dementia because when sCJD is unlikely most positives are false positives. False positive results occur with acute neuronal damage in diverse conditions, including stroke, paraneoplastic disease, inflammation and post-seizure activity.
MR brain imaging characteristically demonstrates hyperintensity of the putamen and caudate head (figure 1A), usually bilaterally. Cortical hyperintensity also occurs (figure 1B). Both sensitivity and specificity based on either bilateral basal ganglia changes or cortical hyperintensity in at least two areas is up to 80%.8 Fluid attenuated inversion recovery and diffusion weighted imaging sequences appear most sensitive, with diffusion weighted imaging being particularly good at detecting early changes in both the striatum and cortex.9 The differential diagnosis for high signal in the basal ganglia includes Wilson's disease, carbon monoxide poisoning, mitochondrial disorders and variant CJD (although in the last the posterior thalamic hyperintensity is more pronounced than the caudate/putamen signal change). However, most of these conditions should be readily clinically distinguishable. The WHO diagnostic criteria for sCJD are currently being modified to incorporate MRI features.8

(A) MR brain scan in sporadic Creutzfeldt–Jacob disease demonstrating hyperintensity of the putamen and caudate heads (arrows). (B) MR brain scan showing cortical hyperintensity (arrows).
The EEG lacks sensitivity in the early stages of sCJD and serial traces may be required to detect the typical pattern
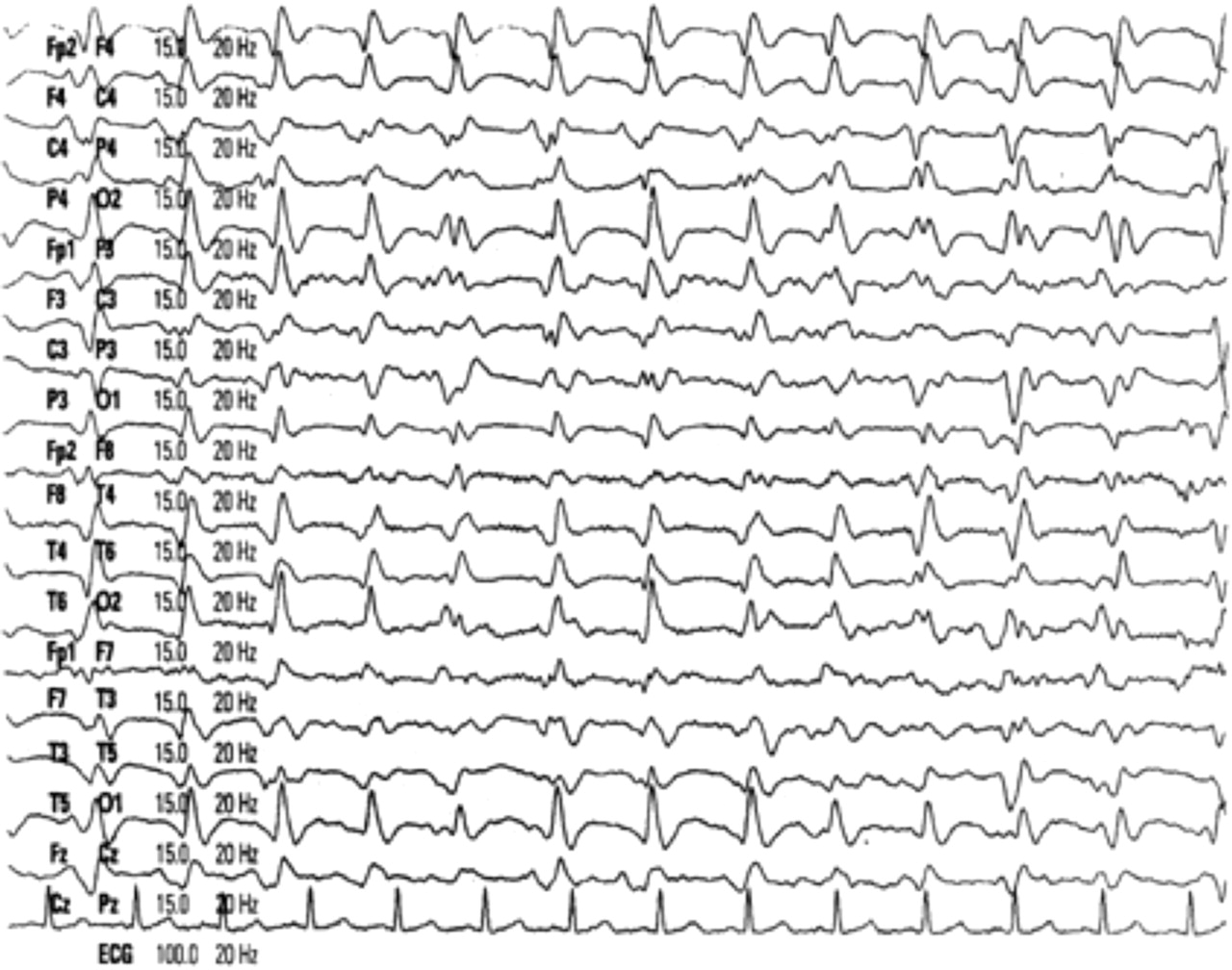
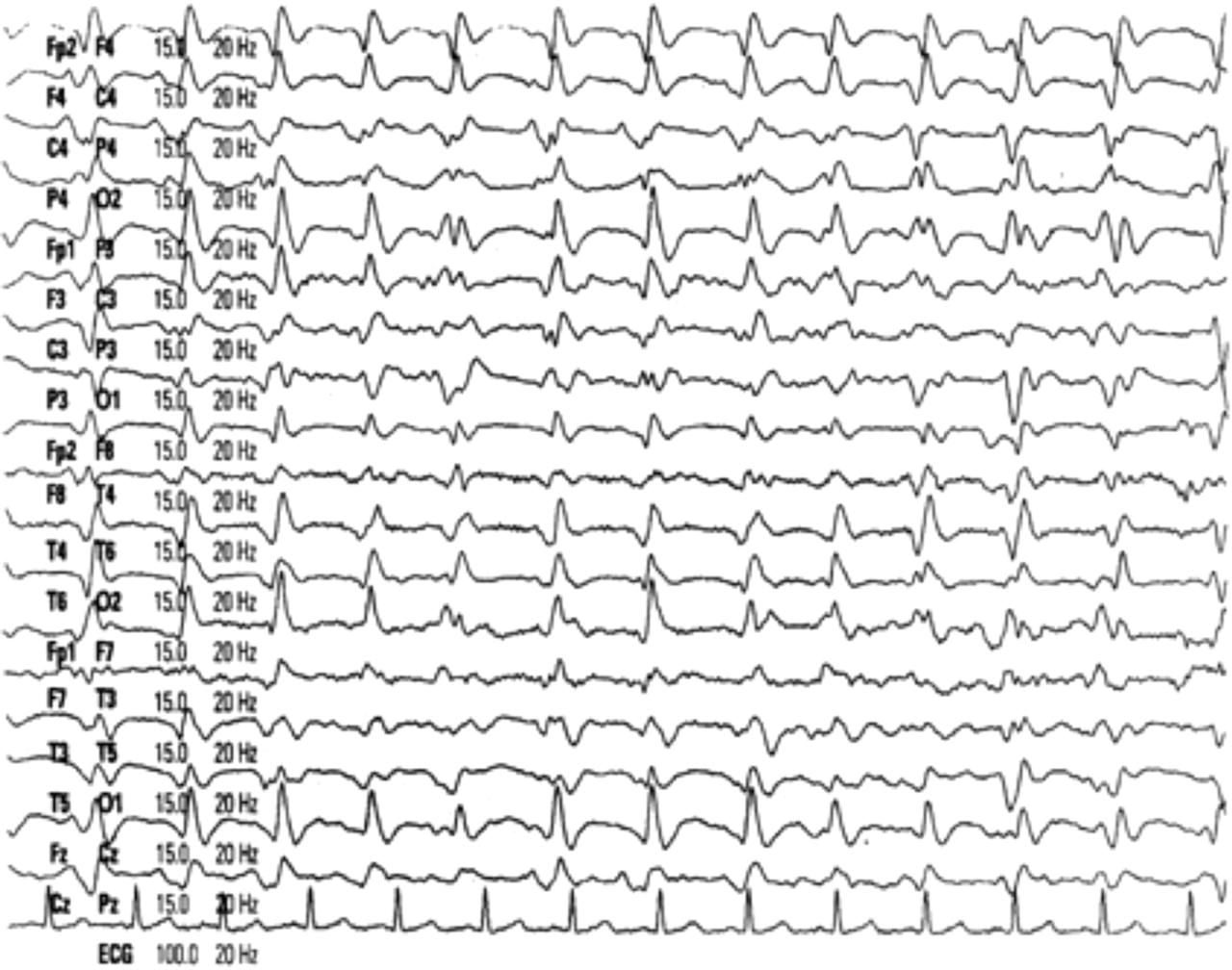
Electroencephalography (EEG) is a useful investigation in sCJD, classically showing periodic, triphasic sharp wave complexes at a frequency of 1/s, usually generalised throughout the trace (figure 2). Objective criteria have been published and when these are applied, the sensitivity of the EEG is about 65% and specificity about 80%.10 However, the EEG lacks sensitivity in the early stages of sCJD and serial traces may be required to detect the typical pattern. False positive EEGs are recognised in Alzheimer's disease, Lewy body dementia, vascular dementia11,–,13 and other less likely to be confusing clinical scenarios, such as lithium toxicity.
{kind=link}
{kind=link}
Typical EEG in sporadic Creutzfeldt–Jacob disease showing generalised periodic triphasic complexes.
sCJD can only be confirmed by pathology but brain biopsy is rarely required in life because of the characteristic clinical phenotype plus supportive investigations (box 1).14
Box 1 Current WHO criteria for sporadic Creutzfeldt–Jacob disease (sCJD): Rotterdam 1998*
Issues in diagnosis
The usual difficulty in diagnosis lies in atypical presentations, or those with negative investigations, in particular cases where the diagnosis may be CJD but there is a nagging doubt that a potentially treatable condition may have been overlooked. No single clinical feature distinguishes CJD. Myoclonus, while typically prominent in CJD, also occurs in many other conditions, including Alzheimer's disease, corticobasal degeneration and immune mediated encephalopathies, so it is not specific. One study found presentation with visual disturbance was the most useful feature distinguishing sCJD from CJD mimics (positive predictive value 93%) but in a highly selected population referred to a CJD unit, whereas extrapyramidal signs and early seizures were relatively more common in non-CJD cases.15
The differential diagnosis of sCJD has proved challenging to study because of referral bias; so much of the literature is based on patients referred to specialised CJD units who are highly selected before they get there and not representative of the average subacute encephalopathy patient in a district general hospital or even in a regional neurology unit. In addition, a number of CJD mimics die with no diagnosis and even autopsy may fail to provide an answer.16 17
Table 2 summarises the most frequent ‘filtered’ sCJD mimics—that is, patients presenting with a rapidly progressive dementia or subacute encephalopathy where readily identifiable alternative diagnoses have been excluded and sCJD is suspected but subsequently disproven.11,–,13 15
‘Filtered’ differential diagnosis of sporadic Creutzfeldt–Jacob disease
In the UK, the two UK CJD surveillance centres perform CSF 14-3-3 testing and genotyping for genetic CJD. All suspected cases of any form of CJD should be discussed with them by faxing the National CJD referral form (downloaded from www.uclh.nhs.uk or www.cjd.ed.ac.uk) to the National CJD Surveillance Unit, Edinburgh and National Prion Clinic, London.
Neurodegenerative sCJD mimics
The commonest sCJD mimics are neurodegenerative, including Alzheimer's disease, dementia with Lewy bodies (DLB), frontotemporal dementia and the tauopathies such as corticobasal degeneration and progressive supranuclear palsy.17 18
Alzheimer's disease
Alzheimer's disease can be mistaken for sCJD for a number of reasons. While an individual with Alzheimer's disease typically survives anywhere between 4 and 8 years, there are more aggressive cases dying after just a year or two. Myoclonus is well recognised and reported in 10%, particularly in the later stages. Investigations can confuse matters, with periodic triphasic waves being seen occasionally on EEG and positive CSF 14-3-3 results.
Dementia with Lewy bodies (DLB)
DLB can also mimic sCJD. The classic DLB phenotype, as defined in the diagnostic criteria, of progressive dementia with particular attentional, visuospatial and subcortical deficits, marked fluctuations, visual hallucinations and parkinsonism is unlikely to be misdiagnosed.19 However, it can present more acutely with rapidly progressive dementia, myoclonus and minimal motor features of parkinsonism. Hallucinations occur in approximately 40% of sCJD patients so this is not a reliable distinguishing feature although in DLB hallucinations are typically visual and well formed, involving animals or people, whereas in CJD they are more variable. EEG in DLB can rarely show periodic complexes, further confusing the issue. However, MRI does not show the typical sCJD caudate/putamen signal change.
Corticobasal degeneration
One study found that corticobasal degeneration was the most frequent single condition masquerading as CJD despite its relative rarity, although it was not population based.17 This probably reflects the selected patient group and frequent occurrence of dementia, myoclonus, alien limb and parkinsonism in corticobasal degeneration (all features well recognised in CJD) in addition to the lack of supportive investigations as corticobasal degeneration is essentially a clinical diagnosis.
Genetic CJD
Genetic CJD should be considered in both typical and atypical sCJD presentations. It refers to autosomal dominantly inherited prion disease with mutations in the prion protein (PRNP) gene. Only 30–50% of inherited cases have a family history, emphasising the need to offer genetic sequencing in all CJD suspects (with informed consent). Depending on the precise mutation, it can present differently to classic sCJD, often in younger individuals with slower disease progression, and sometimes as an alternative clinical phenotype (eg, fatal familial insomnia, Gerstmann–Straussler–Scheinker syndrome).20
Antibody mediated encephalitis
Paraneoplastic encephalitis
Paraneoplastic encephalitis includes limbic encephalitis (typically presenting with memory impairment, behavioural change and seizures) and less frequently brainstem encephalitis. Patients may also have other neurological signs secondary to overlap paraneoplastic antibody effects—for example, a cerebellar syndrome or polyneuropathy. A key feature is the rapidly progressive nature of the neurological syndrome—symptoms evolving slowly over more than a few months are unlikely to be paraneoplastic. The commonest underlying malignancy is small cell lung carcinoma with the individual mounting an antibody response to one or more of their tumour antigens, which then cross react with neuronal antigens. A variety of onconeuronal antibodies have been associated with paraneoplastic encephalitis, including anti-Hu, anti-CV2, anti-Ma and anti-Ta (Ma2), ANNA-3, anti-amphiphysin, anti- N-methyl D-aspartate (NMDA) receptor, anti-glutamate receptor and various other antibodies directed against the neuropil of the hippocampus21:
Anti-Hu is the commonest and these patients often have other, non-limbic neurological features such as ataxia and sensory neuronopathy, in addition to small cell lung cancer.
Anti-amphiphysin syndromes occur with breast cancer and small cell lung cancer, often with stiff person syndrome in conjunction with limbic encephalitis.
Anti-Ma patients are typically middle aged with a range of underlying tumours and frequently a poor neurological response to treatment.
In contrast, anti-Ta (Ma2) classically occurs in young men with testicular germ cell tumours (including extragonadal sites) who respond well to immunotherapy and treatment of the underlying testicular malignancy. 22
Anti-NMDA receptor encephalitis has a well defined phenotype of psychotic encephalopathy, seizures, dyskinesias and autonomic instability with underlying ovarian teratoma. Brain MRI is often normal. It classically presents acutely in young females so is rarely confused with sCJD but it is important as it responds to removal of the teratoma and immunotherapy.23 It is also reported without associated tumour.24
Genetic CJD should be considered in both typical and atypical sCJD presentations
Investigations in paraneoplastic limbic encephalitis are usually abnormal, with brain MRI showing atrophy or mesial temporal signal change in the majority, and CSF pleocytosis and raised protein. However, normal CSF does not exclude the diagnosis. Treatment is aimed primarily at the underlying malignancy, supplemented with immunosuppression in some cases.
It is important to remember that up to 40% of paraneoplastic limbic encephalitis cases have no detectable antibodies with current testing although this figure will shrink as more antibodies are identified. In these cases the diagnosis is based on the clinical phenotype evolving over a maximum of 12 weeks with radiological or pathological evidence of limbic system involvement and discovery of an appropriate cancer.25 As with all paraneoplastic syndromes, discovery of the underlying malignancy frequently postdates the neurology, sometimes by several years. This means that if initial cancer screening investigations (eg, mammography, tumour markers, testicular ultrasound, CT chest/abdomen/pelvis or whole body positron emission tomography depending on availability) are negative they should be repeated after an interval. The frequency and duration of these malignancy screening tests are debated but a pragmatic approach might be after 6–12 months initially, and subsequently annually up to 5 years.
Up to 40% of paraneoplastic limbic encephalitis cases have no detectable antibodies
Voltage gated potassium channel associated limbic encephalitis
This has only recently entered the literature as a sCJD mimic.26 Anti-voltage gated potassium channel (VGKC) antibodies were first recognised in association with limbic encephalitis in 200127 but they are not specific, being associated with the peripheral nerve excitability syndromes of acquired neuromyotonia (Isaac's syndrome) and cramp fasciculation syndrome, in addition to Morvan's syndrome (peripheral and central involvement). In 2004 a review of the clinical and immunological features of 10 cases of VGKC associated limbic encephalitis was published.28 Since then a large number of patients have been identified with an expanding clinical phenotype. Characteristically it presents with a subacute amnesic syndrome plus seizures, but increasingly other presentations are recognised, including sleep disorders (loss of rapid eye movement (REM) sleep, REM sleep behaviour disorder), tremor and refractory epilepsy. Hyponatraemia is common secondary to the syndrome of inappropriate antidiuretic hormone secretion and this may help distinguish it from paraneoplastic limbic encephalitis.
The antibody assay uses binding of radio-iodine labelled α-dendrotoxin to specific potassium channels. The normal reference range is less than 100 picomolar (pM), with limbic encephalitis being associated with titres greater than 400 pM. The significance of intermediate antibody levels (100–400 pM) is unclear. According to one study, just under half the patients have mild lymphocytosis or raised protein in the CSF but oligoclonal bands are rare. The CSF was entirely normal in approximately a quarter.29 Similar to paraneoplastic limbic encephalitis, MRI characteristically reveals temporal lobe signal change but can be normal. EEG abnormalities include focal temporal sharp waves and generalised slowing.25 It is potentially treatable with immunosuppression.
Hashimoto's encephalopathy
This rare CJD mimic is particularly important because, like VGKC encephalitis, it is treatable. It is commoner in women, typically presenting in middle age with a fluctuating encephalopathy including rapid cognitive decline, seizures, neuropsychiatric manifestations and sometimes movement disorders such as tremor or myoclonus, ataxia, stroke-like episodes and even coma.30 31 However, the clinical phenotype is variable.32 Individuals may be euthyroid, hypothyroid or less often hyperthyroid.
The CSF is frequently abnormal but non-specific with modestly raised protein (generally less than 1g/l) and white cell count ranging from normal to a mild lymphocytic pleocytosis (5–30 × 109/mm3). EEG is usually abnormal with generalised slowing or less frequently showing focal slowing, triphasic waves or epileptiform changes. Brain MRI can be normal or demonstrate non-specific white matter changes, which interestingly frequently resolve with treatment, as do the EEG abnormalities.31
The diagnosis is based on an appropriate clinical phenotype with other conditions excluded in addition to raised antithyroid antibodies (antithyroglobulin and thyroid peroxidase antibody, or TPO previously known as antimicrosomal antibody). The difficulty is that antithyroid antibodies are not specific to Hashimoto's encephalopathy and are common in normal elderly people, so if they are raised other mimics must still be excluded.
There is no correlation between antibody level and disease severity or reliable decline with clinical improvement, and none of the implicated antithyroid antibodies has a well defined antigenic target in the brain. Therefore, it is likely that the antibodies are not pathogenic but simply an epiphenomenon reflecting an underlying autoimmune inflammatory state.30 For this reason many believe Hashimoto's encephalopathy should be relabelled ‘steroid responsive encephalopathy associated with autoimmune thyroiditis’. 31 Treatment is with corticosteroids, at least initially, and virtually all patients improve (44 out of 45 in one series).33 Lack of response should prompt review of the diagnosis.
Primary CNS vasculitis
Primary CNS vasculitis is a rare condition, frequently suspected but seldom confirmed. It typically presents in middle age, more commonly in men, with headache, encephalopathy and sometimes strokes. Serological inflammatory markers and autoimmune bloods (antinuclear antibody, extractable nuclear antigens, etc) are usually normal. CSF and brain MRI are often abnormal but non-specific. Cerebral angiography may be helpful but is of low specificity and sensitivity so the diagnosis requires brain and meningeal biopsy. The importance of this rare condition, and one reason it appears so frequently on differential diagnosis lists, is the potential for improvement with immunotherapy (corticosteroids and cyclophosphamide).34
The commonest malignancies misdiagnosed as prion disease are primary CNS lymphoma and intravascular lymphoma
Cerebral amyloid angiopathy related inflammation may represent one subset of CNS vasculitis. It refers to a distinct subtype of amyloid angiopathy that presents with a subacute encephalopathy, headache, seizures and focal neurological deficits, rather than the more common amyloid angiopathy related haemorrhages. MRI reveals white matter changes, sometimes with vasogenic oedema mimicking a space occupying lesion. If biopsied there is evidence of an inflammatory process around vascular deposits of amyloid-β. Unlike other amyloid deposition diseases this is reported to respond to immunosuppression.35
Malignancies
The commonest malignancies misdiagnosed as prion disease are primary CNS lymphoma and intravascular lymphoma, both rare and challenging to diagnose without brain biopsy.
Primary CNS lymphoma comprises high grade, non-Hodgkin B cell-type lymphoma. Immunosuppression is a risk factor but most still occur in immunocompetent individuals. It can present as a progressive focal neurological deficit or as a subacute encephalopathy, particularly in HIV positive patients. CSF cytology is often negative and brain MRI appearances are highly variable but characteristically demonstrate solitary or multifocal contrast enhancing lesions. Previously it was invariably fatal but recent advances in chemoradiotherapy result in prolonged survival in 20–40%.36
Intravascular lymphoma is an even rarer manifestation of non-Hodgkin's lymphoma where the lymphocytes are restricted to the lumen of small and medium sized blood vessels. It typically presents with symptoms secondary to CNS or skin involvement, or with fever of unknown origin, but can involve virtually any organ. Relapsing stroke-like presentations are classical but subacute dementia can occur. Brain MRI may provide a clue to the diagnosis with acute ischaemic lesions in addition to raised serum lactate dehydrogenase, especially if there is clinical or subclinical skin involvement, but the diagnosis is often only made at autopsy.37
Subacute sclerosing panencephalitis
Subacute sclerosing panencephalitis is due to a mutated, slow but persistent form of the wild measles virus. Although rare and usually a paediatric illness it can be seen in young adults and the clinical phenotype of prominent myoclonus, progressive cognitive decline, visual disturbance, and pyramidal and extrapyramidal signs can easily be mistaken for CJD (although the age of onset is against sporadic prion disease). The typical EEG abnormality in subacute sclerosing panencephalitis is periodic triphasic complexes but usually at a lower frequency than in sCJD. The diagnosis is confirmed by raised antimeasles antibody titre in the CSF.38 It is invariably fatal, usually within 1–3 years of onset, with less than 10% having more prolonged survival.
Investigations and making the diagnosis
Box 2 summarises the recommended investigations in subacute encephalopathy patients where sCJD is suspected. Assuming no diagnosis is apparent after preliminary screening bloods and imaging, the secondline investigations include more extensive blood tests, MR brain imaging, CSF examination and usually EEG. The precise sequence of these tests depends on their availability and the clinical scenario in practice, they often occur simultaneously, and frequently a positive result in one area (eg, anti-VGKC antibody or anti-TPO antibody) does not negate the need for other tests (brain MRI, CSF). In most cases a diagnosis is reached but if not there are a number of options.
Box 2 Investigation algorithm
Initial screening tests
Bloods: urea, electrolytes, full blood count, liver function, thyroid function, glucose, C reactive protein, erythrocyte sedimentation rate, vitamin B12, folate
Urinalysis
Chest x ray
CT brain
Secondline investigations
Bloods: ammonia, antithyroid antibodies, paraneoplastic antibody screen, ANA (antinuclear antibody), ENA (extractable nuclear antigens), ANCA (antinuclear cytoplasmic antibody), anticardiolipin antibodies, Borrelia serology, HIV, neurosyphilis screen, angiotensin converting enzyme, lactate dehydrogenase
CSF: cell count, protein, glucose, cytology, oligoclonal bands, viral PCR, 14-3-3
MRI brain (including DWI, FLAIR and contrast examination)
EEG
Thirdline investigations (variably indicated depending on other results)
Cerebral angiography (digital subtraction angiography)
CSF: AAFB (acid and alcohol fast bacilli) stain, antimeasles antibody titre, Whipple's PCR, JC virus PCR
CT chest/abdomen/pelvis or whole body positron emission tomography
Prion protein genotyping
Jejunal biopsy (Whipple's disease)
Brain biopsy
If the neurology team feel sCJD remains likely despite the lack of overwhelmingly supportive investigations then:
The patient can be discussed and referred to one of the two national UK CJD surveillance teams.
Alternatively, a watch and wait approach can be taken, particularly if the patient is elderly with disease duration over 12 months; in such instances the final diagnosis is likely to be neurodegenerative and hence not curable. This approach needs to be discussed carefully with relatives, with regular reviews scheduled. Obviously making a diagnosis remains the aim, if only to guide prognosis and minimise unnecessary procedures.
Thirdly, further investigations can be undertaken, including repeating earlier tests such as the EEG and brain MRI, particularly if the patient has significantly progressed, in addition to considering brain biopsy. This last option carries significant risk, including haemorrhage, infection, permanent focal deficit and seizures. An analysis of 90 biopsies undertaken to investigate dementia where a reversible cause was suspected found 57% were diagnostic (the most common diagnoses being neurodegenerative) but in only 10 of the 90 cases (11%) did the pathology result directly modify treatment.39 Generally, in suspected CJD biopsy is not necessary or recommended, with possible exceptions being younger cases where CNS vasculitis or another inflammatory process is strongly suspected but cannot be confirmed with less invasive investigations. Neurosurgical instruments used in suspected CJD patients should generally be destroyed and not reused.
A fourth option often raised is an empirical trial of corticosteroids. While many argue this is unlikely to cause harm, steroids do have potentially serious adverse effects, and a blind trial raises difficult questions; if there is no prompt improvement how long do you continue for, at what dose and do you try alternatives such as intravenous immunoglobulin (bearing in mind even Hashimoto's encephalopathy does not always respond to steroids immediately and this is especially true for antibody mediated limbic encephalitis)? Even if patients do appear to improve, one still needs a diagnosis to guide future management. Generally, empirical immunosuppression is only recommended if there is a significant chance of a steroid responsive encephalopathy, as suggested by inflammatory CSF, raised antibody titres or serum markers supporting a diagnosis of a steroid responsive condition like Hashimoto's encephalopathy or VGKC encephalitis.
Practice points
Most sCJD mimics are neurodegenerative and rarely is there a treatable alternative; the minority with potentially reversible pathology tend to be younger, with inflammatory CSF or positive serum antibodies.
Disease duration is the most reliable distinguishing feature18; the diagnosis of sCJD should be reviewed in patients surviving over a year and particularly over 2 years.
CSF examination and EEG are useful but relatively non-specific investigations in the work-up for sCJD and its mimics.
MR brain imaging has emerged as an increasingly valuable tool in identifying sCJD, particularly because most of the main differential diagnoses do not show the same caudate/putamen high signal.
A 44-year-old woman was admitted under the neurology department with a 4 week history of rapid cognitive decline and mild gait unsteadiness. Her only history was of classical migraine but immediately prior to presentation she had experienced 5 weeks of persistent migrainous headache with left-sided sensory disturbance. On examination she scored 47/100 on the Addenbrooke's Cognitive Examination with deficits in all domains; she had myoclonic jerks, truncal ataxia and subtle left-sided pyramidal weakness. Initial investigation revealed normal routine bloods, normal MR brain imaging, CSF with 22×109/ mm3 lymphocytes (decreasing to 6 on repeat CSF a week later), protein of 0.62 g/l, normal glucose, red blood cell count and cytology, and positive unpaired oligoclonal bands. EEG showed bursts of frontally predominant slow wave activity. The initial concern was that she had a subacute encephalopathy, possibly sporadic CJD…….
The 44-year-old woman initially described in this article had ongoing fluctuating confusion and developed auditory and visual hallucinations but no other new features. Further investigation revealed a strongly positive anti-La titre of 132 (normal range 0–25) and weakly positive thyroid peroxidase antibody level of 109 (normal range 0–50 IU/ml). All other blood tests were normal or negative, including thyroid function, antinuclear antibodies, anti-VGKC antibody, anti-NMDA receptor antibody and paraneoplastic screen. Based on these results suggesting an immune mediated process and her inflammatory CSF, she was treated with 1 g/day of methyl prednisolone for 3 days followed by oral prednisolone. Within a week there was a marked improvement in her cognition with resolution of the neurological signs although visual misperceptions persisted. A presumptive diagnosis of a steroid responsive encephalopathy was made. She continued to improve on oral prednisolone, deteriorating when the dose dropped to 30 mg daily and improving again when the dose was increased to 40 mg, and is now leading a normal life.
Acknowledgments
Thanks to Robert Will for his helpful suggestions after reading this article, and David Simpson for the case update. This article was reviewed by Geraint Fuller, Gloucester.
References
Other content recommended for you
- Creutzfeldt–Jacob disease mimics, or how to sort out the subacute encephalopathy patient
- CJD mimics and chameleons
- Presenile dementia syndromes: an update on taxonomy and diagnosis
- Fatal insomnia: the elusive prion disease
- Rare histotype of sporadic Creutzfeldt-Jakob disease, clinically suspected as corticobasal degeneration
- Limbic encephalitis: a clinician’s guide
- Patients with Alzheimer's disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease
- Viral and paraneoplastic encephalitis in a patient with liver transplant with unilateral temporoparietal lobe abnormalities: a diagnostic challenge
- Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management
- Amyloid-beta-related angiitis: a treatable rapidly progressive dementia