Article Text

Abstract
A 70-year-old man presented with respiratory distress and proximal muscle weakness shortly after biopsy of a left forearm mass. The biopsy showed giant cell myositis, and serological investigations identified a grossly elevated serum creatine kinase level, suggesting skeletal muscle damage. Serum troponin T was also high, but troponin I was normal. Serum antiacetylcholine receptor antibodies were positive, and imaging showed a thymoma. He recovered well following intravenous immunoglobulin and corticosteroids, and later underwent thymectomy. He is currently in sustained remission, with no clinically detectable myasthenia, but subsequently, developed hypogammaglobulinaemia. Neurologists should remember giant cell myositis/myocarditis can occur in patients who have myasthenia gravis with thymoma, as it is potentially fatal, but may respond to immunosuppression.
- MUSCLE DISEASE
- MYASTHENIA
- NEUROPATHOLOGY, MUSCLE
- NEUROIMMUNOLOGY
- CARDIOLOGY
Statistics from Altmetric.com
Case report
A 70-year-old man presented with increasing tiredness, back pain, dysphagia to solid foods, night sweats and swelling of his left forearm. He had been previously well. Routine blood investigations and an ECG were normal. Chest X-ray showed normal heart size and clear lungs, but lobular enlargement of the right hilum. Ultrasound scan of the left forearm showed multiple ill-defined hypoechoic lesions and a large focal mass in the brachialis muscle with peripheral vascularity. The differential diagnosis was between lymphoma and metastases; two core biopsies were taken. CT scan of chest and abdomen identified a non-compressive 11.6×6.7×3.7 cm soft tissue mass in the anterior mediastinum containing clusters of calcification, consistent with thymoma (figure 1). There was an 8 mm left supraclavicular lymph node and a 7 mm node behind the left pectoralis minor, but no other lymphadenopathy. Abdominal contents were normal apart from multiple simple cysts in the liver.

Large soft tissue mass in the anterior mediastinum with clusters of calcification (arrow): thymoma, lymphoma and metastasis were likely causes. There was no clear fat plane between the pericardium, ascending aorta and this mass and no pericardial, hilar or internal mammary lymphadenopathy.
Two weeks later, he was readmitted with acute dyspnoea, orthopnoea and weakness. On examination, he was tachypnoeic, and could not talk in full sentences. Eye movements were full, and neck flexor/extensor strength was normal. There was proximal muscle weakness, but he was too unwell to be assessed fully for strength or fatiguability. Within hours of admission, he developed respiratory failure, and required assisted ventilation.
Blood investigations showed normal full blood count, renal and liver function tests, serum electrolytes, total protein and albumin, plasma viscosity, calcium, magnesium and phosphate. The following serum levels were elevated: C-reactive protein 54 mg/L (<10), thyroid-stimulating hormone 4.9 mU/L (0.4–5.0), creatine kinase (3956 U/L (24–195), troponin T (1376 ng/L, <13) and lactate dehydrogenase 1096 iu/L (240–490). An infective screen comprising blood, sputum and urine cultures, HIV and syphilis was negative. A paraneoplastic screen was negative for anti-Hu, Purkinje cells, amphiphysin, Yo, MA1, MA2, Ri and CV2/CRMP5. An autoimmune screen showed normal complement 3 and 4 levels, negative antinuclear antibodies, extractable nuclear antigens, antineutrophil cytoplasmic antibodies, double-stranded DNA and serum protein electrophoresis, but was positive for antiskeletal muscle antibodies and acetylcholine receptor antibodies at 306×10−10 mol (0–5×10−10).
He received 5 days of intravenous immunoglobulin (0.4 g/kg/day) and a 3-day course of intravenous methylprednisolone (1 g daily) followed by a maintenance dose of 40 mg prednisolone daily. Despite the very high serum troponin T, an echocardiogram showed no features of myocarditis and an ejection fraction of 50%–55%. His serum troponin T level dropped to 566 ng/L by day 3 of treatment and to 187 ng/L by day 5. ECG now showed intermittent supraventricular tachycardia and poor R-wave progression. Serum creatine kinase on day 9 was normal (123 iu/L).
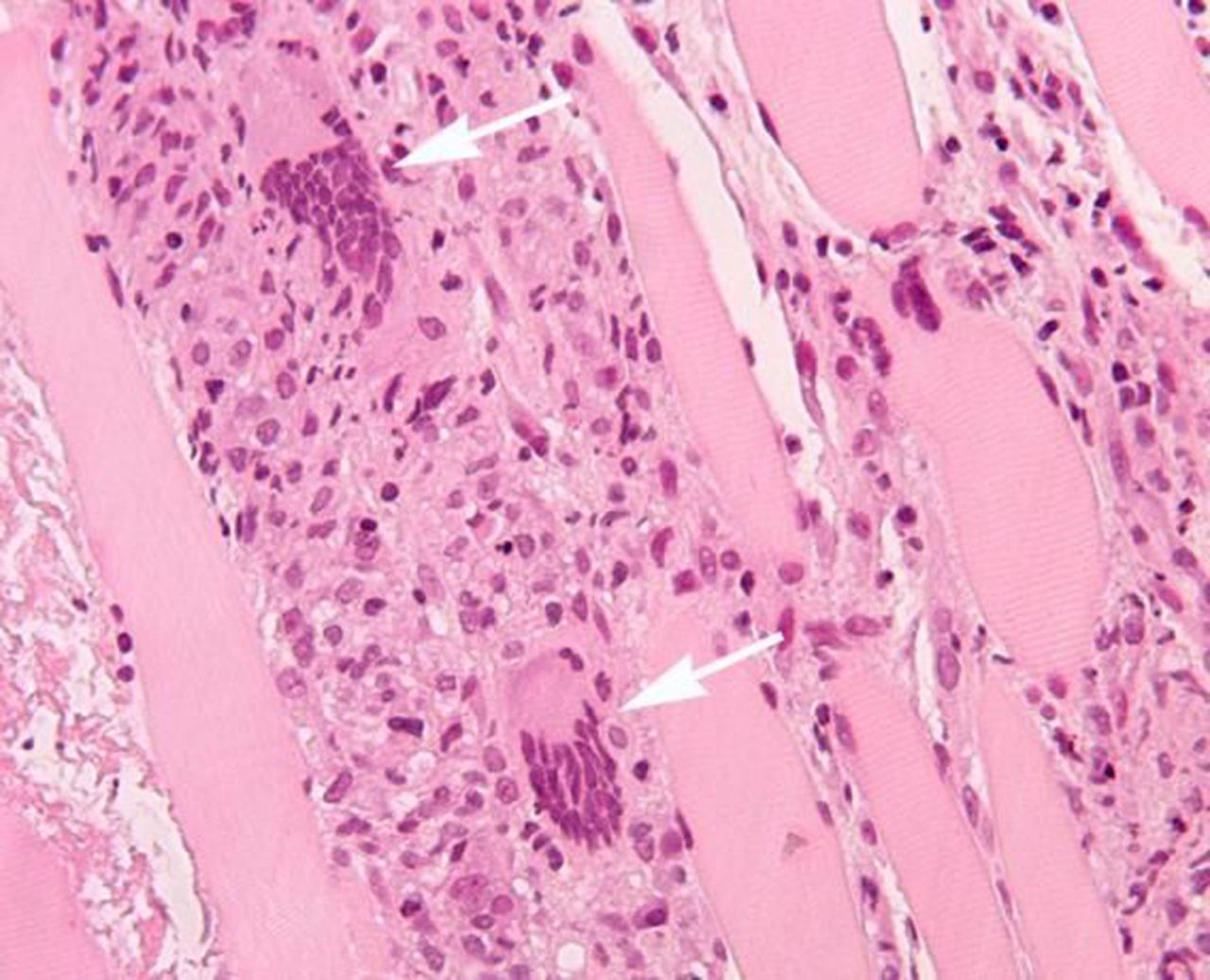
Histopathology from the forearm muscle biopsies showed giant cell myositis (figure 2). A planned CT-guided biopsy of the thymic mass was abandoned—due to risk of pneumothorax—when repeat CT showed the thymic mass was significantly smaller. We were unsure whether his respiratory compromise was due to inflammatory myositis of the respiratory muscles or to uncontrolled myasthenia. A trial of pyridostigmine did not help. Nerve conduction studies showed no demyelinating nerve damage, but the neurophysiologist could not assess for a neuromuscular junction disorder.
{kind=link}
{kind=link}
Forearm biopsy showing florid skeletal muscle myositis, with giant cells (arrows).
His muscle strength gradually recovered, but respiratory function improved only slowly, initially requiring intermittent non-invasive ventilation post decannulation. Repeat CT scan of chest 4 weeks after admission showed the thymic mass was again smaller. Eventually, he was discharged on a reducing dose of prednisolone.
Three months later, he was asymptomatic on prednisolone 20 mg daily, with sufficient recovery to lead a local rambling group, and with no clinical evidence of myasthenia or myopathy. Serial troponin T remained high (50–70 ng/L), and cardiac MR scan 5 months after admission was normal. Eight months after his initial admission, he underwent elective thymectomy. This showed a benign thymoma, histological type AB. One month after thymectomy, he was found to have hypogammaglobulinaemia with low serum IgG and IgM levels. His serum antiacetylcholine receptor antibody titre had dropped to a lower, but persistently elevated, titre (116×10−10 mol).
Discussion
Thymomas express neuronal and muscle autoantigens that result in the formation of autoreactive antibodies. Such autoantibodies may be clinically silent,1 as suggested here by the apparent absence of myasthenia on recovery. Alternatively, they may cause neurological conditions, including myasthenia gravis, myositis, neuromyotonia, encephalitis, subacute hearing loss and autonomic neuropathy.1 There are numerous non-neurological conditions associated with both thymoma (table 1) and myasthenia gravis (box 1). It is important that clinicians make a careful systemic examination on initial assessment and follow-up.
Conditions associated with thymoma
Associations with myasthenia gravis
Rheumatological/Systemic
Rheumatoid arthritis
Systemic lupus erythematosus
Graft-vs.-host disease
Haematological
Pernicious anaemia
Neurological
Neuromyelitis optica
Thymoma, myositis and myasthenia gravis may occur together. The largest published series by Suzuki et al2 found that 0.9% of patients with myasthenia gravis develop inflammatory myopathies. Cardiac muscle may also be affected, although less commonly (three of eight described cases). Patients with myasthenia gravis who develop myositis are more likely to express striational antibodies; certain subtypes (especially antiryanodine receptor and anti-VGKC Kv1.4) suggest more severe disease.3 Myositis may either occur concurrently with, or precede the development of, myasthenia gravis.4 Conversely, myocarditis seems always to follow myasthenia gravis, with a widely variable delay of 13–211 months.2 The pathological subtype of myositis in this case was giant cell (granulomatous myositis) as previously reported.2 Giant cell myositis may also occur in sarcoidosis, lymphoma, infectious diseases (tuberculosis, syphilis, Pneumocystis jirovecii) and inflammatory bowel disease.5
The presence and extent of cardiac involvement in our patient remains unclear. While serum troponin T elevation and ECG abnormalities suggested significant myocardial involvement, his echocardiogram was normal, and he never developed cardiac compromise. One possible explanation is that he had already started treatment by the time of the echocardiogram. Troponin T may possibly be falsely elevated (false positive) in myositis due to cross reactivity in the assay with skeletal troponin T.6 His troponin I level was normal when tested after thymectomy despite persistently elevated troponin T (73 ng/L). It is not clear whether troponin T is more sensitive or less specific than troponin I.
There is no standard approach to treating myasthenia gravis complicated by myositis. However, it is critically important to start immunotherapy immediately upon suspicion, as the associated myocarditis is often rapidly fatal due to lethal arrhythmias and congestive heart failure.7 ,8 Myasthenia gravis with myositis and cardiac involvement can respond well to oral and intravenous corticosteroids, tacrolimus and plasma exchange;2 ,9 the therapeutic choice can be driven by the presence of myasthenic crisis. Escalating doses of pyridostigmine require caution as high doses may worsen weakness due to cholinergic crisis; pyridostigmine may also be arrhythmogenic, and increase bronchial secretions,10 particularly important with coexisting myocarditis.
Our patient responded well and rapidly to treatment. This appeared partly due to starting immune treatment rapidly, enabled by having the imaging and histology results by the time he developed respiratory distress.
Hypogammaglobulinaemia may sometimes accompany thymoma (Good syndrome);11 it may persist after thymectomy, suggesting no direct relation to the tumour, but possibly a related autoimmune phenomenon.12 Clearly, this increases the risk of infection, and leads to difficult treatment decisions regarding immunosuppression, especially if there are no myasthenic or myositic symptoms. We do not know if the serum immunoglobulin level was low in this patient earlier in the course of his illness.
Giant cell myocarditis may potentially cause sudden death in patients with myasthenia gravis, especially if there is a thymoma.13 In addition to highlighting the wide variety of neurological and non-neurological disorders that accompany thymoma, this case highlights how early identification of giant cell myositis in patients with myasthenia gravis may critically influence treatment decisions and outcome.
Key points
Giant cell myositis/myocarditis and myasthenia gravis may occur together in association with thymoma.
Raised serum creatine kinase and troponin levels in myasthenia suggest myositis and myocarditis; striational antibodies may be positive.
Myositis/myocarditis in myasthenia gravis is frequently fatal, but can be successfully treated with immunosuppression.
Thymoma has several other neurological and non-neurological associations, including hypogammaglobulinaemia.
Serum levels of troponin T and troponin I may show disparity, complicating the diagnosis of myocarditis.
Footnotes
Contributors AS: drafting of the manuscript, initial patient assessment. AP: drafting of the manuscript, literature review. SW: attending consultant neurologist looking after patient, initial patient management, critical review of manuscript. DH: pathological analysis of muscle biopsy, provision of pathology images and differentials of giant cell myositis, critical review of manuscript. EH: consultant neurologist overseeing patient care, critical review of manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed. This paper was reviewed by Jon Walters, Swansea, UK.
Other content recommended for you
- Morvan’s fibrillary chorea: a paraneoplastic manifestation of thymoma
- Giant cell myositis associated with metastatic thymoma and granulomatous hypercalcaemia
- Neuromyelitis optica and myasthenia gravis in a young Nigerian girl
- Clinical characteristics, time course, treatment and outcomes of patients with immune checkpoint inhibitor-associated myocarditis
- 68Ga-DOTATOC PET/CT to detect immune checkpoint inhibitor-related myocarditis
- Immune checkpoint inhibitor-mediated myasthenia gravis with focal subclinical myocarditis progressing to symptomatic cardiac disease
- Overlooked non-motor symptoms in myasthenia gravis
- Consensus disease definitions for neurologic immune-related adverse events of immune checkpoint inhibitors
- Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events
- Fulminant immune-mediated necrotising myopathy (IMNM) mimicking myocardial infarction with non-obstructive coronary arteries (MINOCA)