Article Text

Abstract
Background Different amyotrophic lateral sclerosis (ALS) phenotypes have been recognised, marked by a varying involvement of spinal and bulbar upper and lower motor neurons. However, the differential characteristics of these phenotypes are still largely unknown.
Objective To define the epidemiology and outcome of ALS phenotypes in a population based setting.
Methods All ALS cases incident in two Italian regions were prospectively collected from 1995 to 2004 in an epidemiological register. Cases were classified according to established ALS phenotypes: classic, bulbar, flail arm, flail leg, pyramidal, respiratory, pure lower motor neuron (PLMN) and pure upper motor neuron (PUMN).
Results ALS phenotype were determined in 1332 out of 1351 incident patients (98.6%). Classic and bulbar phenotypes had similar mean annual incidence rates. Gender specific incidence rates showed a male preponderance in respiratory, flail arm, classic and PLMN phenotypes; in all other phenotypes, men and women had similar incidence rates. Age at onset was significantly lower in pyramidal, PLMN and PUMN phenotypes and higher in the bulbar phenotype. The best outcomes were observed in PUMN, pyramidal, PLMN and flail arm phenotypes and the worst in respiratory and bulbar phenotypes.
Conclusions Our epidemiological findings suggest that ALS phenotypes carry distinctive and easily distinguishable clinical and prognostic characteristics, strongly related to a complex interplay between gender and age. The categorisation of ALS patients according to more homogenous clinical groups is relevant in identifying biological markers for ALS and should be considered for the design of clinical trials.
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder of adult life characterised by the progressive involvement of lower and upper motor neurons at the bulbar and spinal level. In 5–10% of patients, a positive family history for ALS can be detected. However, in most patients the cause of ALS remains unknown. It is a generally accepted notion that the clinical spectrum of ALS includes different phenotypes marked by a varying involvement of spinal and bulbar upper and lower motor neurons.1 2 Accordingly, eight distinctive clinical phenotypes are recognised in the literature: classic, bulbar, flail arm, flail leg, pyramidal, respiratory, pure lower motor neuron (PLMN) and pure upper motor neuron (PUMN).3–9
A discussion has recently arisen concerning the possibility that different ALS presentations have different aetiologies or underlying factors—whether genetic, environmental or both—that modify the phenotype.10 However, at present, no studies have been carried out to assess and compare all ALS phenotypes using an epidemiological approach.
The aim of this study was to evaluate the clinical characteristics and outcome of different ALS phenotypes in a large population based setting.
Methods
The Piemonte and Valle d'Aosta Register for ALS (PARALS) is a prospective register collecting all cases of ALS in the Piemonte and Valle d'Aosta regions of Italy (total population at the 2001 national census 4 332 842; total area 28 692 km2). The register was established in 1995 and is still in operation. Epidemiological data regarding the 1995–2004 period have recently been published.11
Case collection
The main sources of cases were the neurology departments of the two regions. Investigators used an ad hoc questionnaire to collect patient demographic data, disease history, neurological and laboratory findings, and treatments. Diagnostic EMG examination was performed in all patients according to standard procedures. The secondary sources for case collection were: the Piemonte and Valle d'Aosta Central Regional Archives; and the mortality coding from the Italian Bureau of Statistics. Clinical records of cases found through secondary sources were obtained, and relevant clinical information for each case was analysed in order to verify if the patient met the eligibility criteria; all living patients were contacted by phone and visited by one of the neurologists involved in the study.
Diagnostic criteria
The diagnosis of ALS was based on the original El Escorial diagnostic criteria12 although from 2000, cases were also classified according to the El Escorial revised criteria.13 Patients with PUMN4 5 and PLMN6 were prospectively included in the register; they were not considered in the original epidemiological paper11 but are included in this study. A clinical follow-up of each patient was performed at regular intervals (2–4 months). A standard form was used for collecting clinical information at each follow-up visit. The presence of frontotemporal dementia (FTD) was determined using an internally generated questionnaire administered to caregivers during the follow-up visits and was based on Neary's criteria.14 15
Phenotypic classification
We classified the patients into the eight recognised phenotypes of ALS: classic, bulbar, flail arm, flail leg, pyramidal, respiratory, PLMN and PUMN. The classification was based on clinical data gleaned from all available sources (clinical charts, clinical notes of the collaborating centres, including the standard forms used for the register) which were prospectively collected during patient follow-up. It was established according to the clinical and EMG picture at diagnosis and revised during the follow-up.
Classic (Charcot's) phenotype
Classic ALS was characterised by onset of symptoms in the upper or lower limbs, with clear but not predominant pyramidal signs.
Bulbar phenotype
These patients had a bulbar onset with dysarthria and/or dysphagia, tongue wasting, fasciculation and no peripheral spinal involvement for the first 6 months after symptoms onset. Pyramidal signs were not required to be evident in the first 6 months but needed to be evident thereafter.
Flail arm phenotype
Patients in this group were characterised by progressive, predominantly proximal, weakness and wasting in the upper limbs. In this category we also included patients with pathological deep tendon reflexes or Hoffman sign in the upper limbs at some point during the disease but without hypertonia or clonus. Functional involvement had to be confined to the flail limbs for at least 12 months after symptom onset.3
Flail leg phenotype
Patients were characterised by progressive distal onset of weakness and wasting in the lower limbs. In this category we also included patients with pathological deep tendon reflexes or Babinski sign in the lower limbs at some point during the disease but without hypertonia or clonus. Patients with wasting and weakness beginning proximally in the legs without distal involvement at presentation were classified as classic ALS.3
Pyramidal phenotype (predominant upper motor neuron ALS)
These patients had clinical manifestations dominated by pyramidal signs, mainly severe spastic para/tetraparesis, associated with one or more of the following signs: Babinski or Hoffmann sign, hyperactive reflexes, clonic jaw jerk, dysarthric speech and pseudobulbar affect. Spastic paresis could be present at the beginning or in the fully developed stage of the disease. These patients showed at the same time clearcut signs of lower motor neuron impairment from onset of the disease, as indicated by muscle weakness and wasting and by the presence of chronic and active denervation at the EMG examination in at least two different sites.7 8
Respiratory phenotype
These patients had prevalent respiratory impairment at onset, defined as orthopnoea or dyspnoea at rest or during exertion, with only mild spinal or bulbar signs in the first 6 months after onset. These patients showed signs of upper motor neuron involvement.9
Pure lower motor neuron
These patients had clinical and electrophysiological evidence of progressive LMN involvement. From this category we excluded patients with motor conduction block(s) on extensive standardised nerve conduction studies, clinical UMN signs, history of disease that mimics motor neuron disease, family history of inherited spinal muscular atrophy and deletion in the SMN1 gene or expansion of CAG repeat in the androgen receptor gene. Neuroimaging studies were performed to rule out structural lesions.6
Pure upper motor neuron
These patients had clinical signs of UMN involvement—that is, severe spastic para/tetraparesis, Babinski or Hoffmann sign, hyperactive reflexes, clonic jaw jerk, dysarthric speech and pseudobulbar affect. From this category we excluded patients with clinical or EMG signs of LMN involvement, according to the El Escorial criteria, during the follow-up, and those with a history of disease that mimics motor neuron disease, family history of spastic paraparesis/tetraparesis and mutation of genes related to hereditary spastic paraplegia (SPG3A, SPG4, SPG6, SPG7 and SPG20).4 5
Statistical methods
Ninety five per cent CIs were calculated assuming a Poisson distribution.16 Comparison between means were made with analysis of variance. The effect of age and gender on ALS phenotypes was assessed with multivariate analysis of variance, including age, gender and the interaction between age and gender as dependent variables. Survival was calculated to death/tracheostomy or censoring date (31 December 2009), using the Kaplan–Meier method, and compared with the log rank test. Multivariable analysis was performed with Cox's proportional hazards model (stepwise forward) (see supplementary table 2, available online, for a list of the variables included in the model).
A p value <0.05 was considered significant. Data were processed using SAS statistical package (V.8.2). No patient was lost to follow-up.
Standard protocol approvals, registrations and patient consent
The study design was approved by the ethics committee of the coordinating centre. Patient consent was not required since this study did not modify the routine clinical practice. However, databases were managed according to the Italian law for the protection of privacy.
Results
A total of 1351 patients were diagnosed with ALS in the period 1995–2004 in Piemonte and Valle d'Aosta.11 Clinical phenotype was established in 1332 patients (98.6%). Clinical phenotype was established blindly by three of the authors (AC, LM, GM). When the classification differed, a discussion was undertaken to reach a consensus. In 19 patients (1.4%), no consensus was reached, mainly due to lack of full clinical details; these cases were excluded from the analysis. These patients did not differ in terms of demographic or clinical characteristics from those with full clinical details.
Epidemiological and clinical characteristics of ALS phenotypes
The clinical features of ALS phenotypes are reported in table 1 and their mean annual incidence rates and gender rate ratios are shown in table 2.
Mean age at onset, mean time delay from onset to diagnosis and frequency of frontotemporal dementia
Amyotrophic lateral sclerosis phenotypes. Overall and men versus women mean annual crude incidence rates (/100 000 population), 95% CIs and gender incidence rate ratios
Classic phenotype. This is the commonest ALS phenotype in men and the second commonest in women, with a male to female rate ratio of 1.65:1. Age at onset is 62.8 years (SD 11.3), with a peak of the age specific incidence rate in the seventh decade in both genders (figure 1A). Four per cent of cases with this phenotype have FTD. Median survival time is 2.6 years (see supplementary table 1 available online only), with a 10 year survival rate of 13.0%.
Bulbar phenotype. Bulbar ALS has the same incidence in the two genders (1/100 000 population), with a male to female rate ratio of 0.98:1. The peak of the age specific incidence rate is in the eighth decade in both genders (figure 1B). Nine per cent of bulbar patients have FTD, the highest figure among the ALS phenotypes. Median survival time of bulbar ALS is the second worst (2.0 years); only 3.4% of patients survived up to 10 years.
Flail arm phenotype. This phenotype is relatively rare and more common in men (incidence rates 0.28 in men and 0.07 in women), with a male to female rate ratio of 4.00:1. Mean age at onset is 62.6 years (SD 11.8). In this phenotype, FTD is very rare (1.4%). Flail arm phenotype is relatively benign, with a median survival time of 4.0 years and a 10 year survival rate of 17.4%. In these patients, ALS remains restricted to the upper limbs for a mean of 20 months after onset.
Flail leg phenotype. This phenotype has a similar incidence in the two genders, with a male to female rate ratio of 1.03:1. Mean age at onset is the second highest (65.0 years) and the peak of age specific incidence rate is in the eighth decade (figure 1D). FTD is present in 4% of patients with this phenotype. Median survival time (3.0 years) and the 10 year survival rate (12.8%) of this phenotype are similar to those of classic ALS. In these patients, ALS remains restricted to the upper limbs for a mean of 16 months after onset.
Pyramidal phenotype. Patients with this phenotype have a quite young age at onset (58.3 years). Both genders are equally represented, with a male to female rate ratio of 1.04:1. The peak of the age specific incidence rate is in the seventh decade (figure 1E). FTD is rather uncommon (2.5%). Median survival time is 6.3 years and the 10 year survival rate is 31.9% (see supplementary table 1 available online).
Respiratory phenotype. This is the rarest phenotype (annual incidence rate: men 0.06/100 000; women 0.01/100 000). Median survival time is 1.4 years and no patient with this phenotype survived up to 10 years.
PLMN. PLMN has quite a low incidence rate and is twice as frequent in men (male to female rate ratio 2.04:1). Patients with PLMN are younger than those with any other ALS phenotype (56.2 years), with a peak of the age specific incidence rate in the seventh decade among men and in the sixth decade among women (figure 1F). No patient with the PLMN phenotype developed FTD. PLMN patients have a longer survival than any other phenotype besides PUMN (median survival time 7.3 years).
PUMN. PUMN has a relatively low incidence rate (0.12 in both genders) and mean age at onset (58.9 years). The peak of age specific incidence rate is in the sixth decade in both genders (figure 1G). Median survival time of PUMN is the longest among ALS phenotypes (13.1 years), with a 10 year survival rate of 71.1%.

Amyotrophic lateral sclerosis (ALS) phenotypes: incidence rates according to age group for males versus females. Respiratory phenotype is not shown due to the low number of cases. PLMN, pure lower motor neuron phenotype; PUMN, pure upper motor neuron phenotype. (A) Classic ALS; (B) Bulbar ALS; (C) Flail arm phenotype; (D) Flail leg phenotype; (E) Pyramidal phenotype; (F) PLMN; (G) PUMN.
Relative frequency of ALS phenotypes through the age groups
The relative frequency of ALS phenotypes showed an intriguing trend with age: among men (figure 2A), there was a decline in frequency of flail arm, pyramidal and classic phenotypes with increasing age, and an increase in bulbar (from 10% to 51%) and flail leg (from 0 to 12%) phenotypes; among women (figure 2B), the decline in frequency of pyramidal (from 37.5% to 6%) and classic (from 37.5% to 12%) phenotypes was even more pronounced, as well as the rise in frequency of the bulbar phenotype (from 6% to 72%).

Relative frequency of amyotrophic lateral sclerosis phenotypes according to age. (A) Men; (B) women. The percentage is calculated on the incidence rates. PLMN, pure lower motor neuron phenotype; PUMN, pure upper motor neuron phenotype.
Influence of age and gender on ALS phenotypes
According to the results of the multivariate analysis of variance, 54% of total variance of ALS phenotypes was explained by age and gender.
ALS phenotypes survival
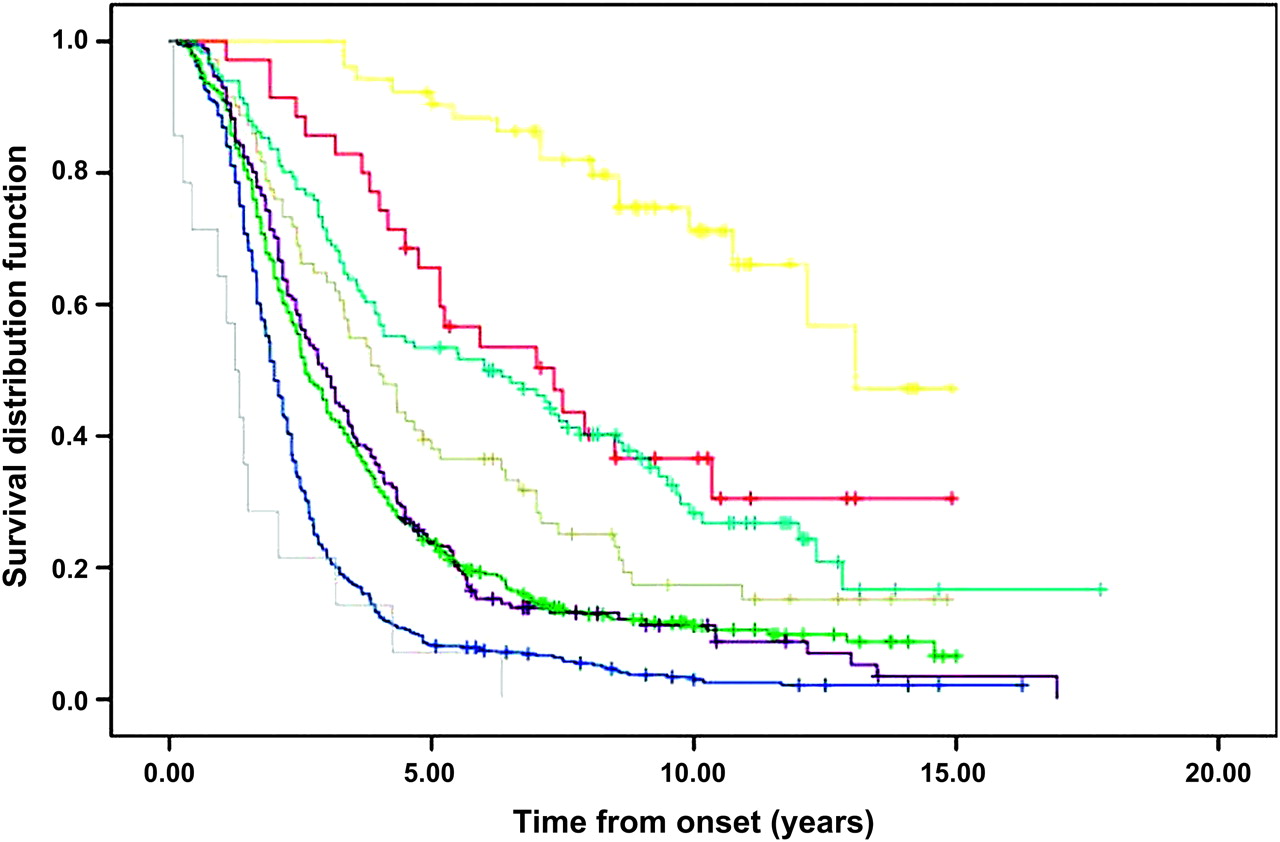
The survival curves of ALS phenotypes are compared in figure 3. The outcome of the different phenotypes differed significantly (p<0.0001). At multivariable analysis (Cox model) (see supplementary table 2 available online only), the variables independently related to outcome were age at onset (p<0.0001), bulbar phenotype (p<0.0001), PUMN (p<0.0001), pyramidal phenotype (p<0.0001), respiratory phenotype (p<0.0001), FTD (p=0.0008) and flail arm phenotype (p=0.008).

{kind=link}
{kind=link}
{kind=link}
Tracheostomy free survival, according to amyotrophic lateral sclerosis (ALS) phenotype. Yellow, PUMN; red, PLMN; light blue, pyramidal ALS; grey, flail arm; violet, classic ALS; green, flail leg; blue, bulbar; cyan, respiratory. Crosses are censored patients. PLMN, pure lower motor neuron phenotype; PUMN, pure upper motor neuron phenotype.
Discussion
This is the first comprehensive survey directly comparing all ALS phenotypes in a large epidemiological setting with a prospective design. The advantage of this large population based study is that it includes detailed and standardised phenotypic information, thus avoiding the referral bias that is inherent to clinic based cohorts.17
The main finding of our study is that ALS clinical phenotypes carry highly distinctive clinical, demographic and prognostic characteristics. In brief, the bulbar phenotype is typical of older patients, has similar incidence rates in the two genders and carries the worst survival. At the opposite extreme, the pyramidal phenotype and PUMN are typical of younger patients and have the most benign outcome. Flail arm phenotype is rare, more frequent in men, and has a relatively good prognosis, while flail leg phenotype, which is the third more common phenotype, has an equal incidence in both genders and a slightly worse outcome. The classic phenotype has the highest incidence among men, and carries an intermediate outcome.
The different clinical and demographic characteristics of ALS phenotypes translate into different probabilities of having a presentation with a specific phenotype at different ages. As shown in figure 2, the bulbar phenotype represented less than 10% of cases in the under 39 year age group, and increased up to more than 50% in those aged more than 80 years, with a more marked trend among women, whereas the classic phenotype steadily decreased, going from the younger to the older decades. The influence of age on ALS clinical features has also been reported in a clinical based series of ALS patients in Japan.18
The male predominance generally reported in ALS17 19 is true for only four phenotypes—classic, flail arm, respiratory and PLMN. The two genders were almost equally represented among the other phenotypes. Therefore, when considering incidence rates and not absolute figures, the predominance of women in the bulbar phenotype described in clinical series appears to be simply related to the older age at onset of this phenotype.
ALS phenotype bears a strong influence on disease outcome. Bulbar and respiratory phenotypes carried the worst prognosis20 while the pyramidal phenotype had by far the longest survival.7 Interestingly, the survival time of the pyramidal phenotype of ALS was shorter than that of PUMN, but longer than all of the other ALS phenotypes, supporting the notion that it represents an intermediate form between classic ALS and PUMN.8 21 Also, the flail arm phenotype had a relatively good outcome, with a median survival time of 4 years.3 22 The outcome of classic and flail leg phenotypes were similar, at variance with the findings of a previous paper reporting a better prognosis for the flail leg phenotype.3 Cox's multivariable analysis confirmed the independent role of ALS phenotype on survival and suggests that this classification should be used for patient stratification in clinical trials—for example, utilising the minimisation method.23
FTD was present in about 5% of patients, mainly those with bulbar phenotype. However, many ALS patients have evidence of frontotemporal behavioural dysfunction that may not satisfy Neary criteria for FTD and subclinical syndromes have not been considered in the present paper.24
Our findings support the idea that ALS is a clinically heterogeneous disease. The heterogeneity of motor phenotypes in ALS has been recently analysed considering three symptom dimensions: body region of onset, relative mix of upper motor neuron and lower motor neuron involvement and rate of progression,2 and hypothesising that the initial trigger of ALS is a stochastic process, with an apparently random but focal initiation. According to our findings, however, the focal initiation of ALS is not entirely random as it is strongly determined by patient age and gender, as indicated by the fact that these two factors explained about 50% of the clinical variance.
A previous study analysed ALS prognostic groups using a latent class clustering.25 The authors identified five classes, of which the first included most patients with classic, flail arm and flail leg phenotypes, the second, which carries the worst prognosis, includes most bulbar onset patients, and the fourth and fifth include patients with mixed phenotypes but with a milder clinical course.
The reasons of the influence of these two factors on ALS clinical presentation remain unknown at present. In the preclinical model of ALS, the SOD1 transgenic mouse, male rodents carrying the human mutated gene had an earlier age at onset, an early progress of locomotor rating scores and a shorter survival than their female counterparts, with a clear gender related phenotypic dimorphism.26 27 This dimorphism has been related to gonadal hormones.28 However, in human ALS, data on reproductive factors and sexual hormones are scarce and contradictory,29 30 not allowing any definitive conclusions to be drawn.
A strong effect of age and gender on clinical phenotype has also been reported in Parkinson's disease31 where the later age at onset, the more frequent presentation with tremor and the milder clinical course in women have been explained by the higher initial dopamine levels in the substantia nigra, which delay the moment of reaching a critical threshold of striatal dopamine depletion. Unfortunately, in ALS there are no studies assessing the age and gender related diversities in the motor neuron pool and the receptor function at the upper and lower motor neuron level.
Genetic factors may play a role in determining the range of ALS phenotypes although to date no genes have been shown to have a definite effect on phenotype. Patients with SOD1 mutations show a phenotypic heterogeneity even within the same mutation32 although some specific missense mutations carry a consistently worse (ie, A4V, G41S) or better (ie, H46R, G93C) prognosis. Patients with FUS mutations are also quite heterogeneous but some mutations seem to carry more defined phenotypes: the R514S and R521C missense mutations are characterised by a predominantly proximal and axial phenotype33 34 and the P525L missense mutation is characterised by a very young age at onset (<30 years), with a bulbar presentation and a short duration.33 35
The role of environmental factors on ALS phenotypic expression cannot be ruled out. The overall higher frequency of ALS in men could be related to occupational exposures to metals and toxins, while the increase in cigarette smoking, an established exogenous risk factor for ALS,36 could explain the progressive reduction of the gender gap reported in recent ALS epidemiological studies.17
We conclude that our epidemiological data strongly support the fact that recognised ALS phenotypes have different clinical, demographic and outcome characteristics, and can be recognisable even in a population based setting. The pathophysiological bases of the varying pattern of motor neuron degeneration, partly related to age and gender, are still largely unknown. A better understanding of the factors that influence the phenotypic expression of ALS would be important for identifying the underlying biochemical, genetic and environmental mechanisms of the disease. Moreover, the categorisation of ALS patients according to clinical phenotypes should be considered for improving the design of clinical trials and to better focus the communication of diagnosis and outcome with patients and their families.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
See Editorial commentary, p 711
↵* The other members of the PARALS study group are: R Mutani, MD (Department of Neuroscience, University of Torino, Advisory Committee); M Balma, MD (Department of Neuroscience, University of Torino, site investigator); S Cammarosano, MD (Department of Neuroscience, University of Torino, site investigator); A Canosa, MD (Department of Neuroscience, University of Torino, site investigator); S Gallo, MD (Department of Neuroscience, University of Torino, site investigator); A Ilardi, MD (Department of Neuroscience, University of Torino, site investigator); L Durelli, MD (Department of Neurology, University of Torino and AOU San Luigi Gonzaga, Orbassano, Advisory Committee); B Ferrero, MD (Department of Neurology, University of Torino and AOU San Luigi Gonzaga, Orbassano, site investigator); S De Mercanti, MD (Department of Neurology, University of Torino and AOU San Luigi Gonzaga, Orbassano, site investigator); A Mauro, MD (Department of Neurorehabilitation, University of Torino, Istituto Auxologico Italiano, IRCCS, Piancavallo, Advisory Committee); M Leone, MD (Department of Neurology, University of Piemonte Orientale ‘Amedeo Avogadro’, and AOU Maggiore, Novara, Advisory Committee); F Monaco, MD (Department of Neurology, University of Piemonte Orientale ‘Amedeo Avogadro’, and AOU Maggiore, Novara, Advisory Committee), N Nasuelli, MD (Department of Neurology, University of Piemonte Orientale Amedeo Avogadro, and AOU Maggiore, Novara, site investigator); L Sosso, MD (Department of Neurology, Ospedale Mauriziano, Torino, site investigator); M Gionco, MD (Department of Neurology, Ospedale Mauriziano, Torino, site investigator); A Marchet, MD (Department of Neurology, Ospedale Martini, Torino, site investigator); C Buffa, MD (Department of Neurology, Ospedale Maria Vittoria, Torino, Advisory Committee); R Cavallo, MD (Department of Neurology, Ospedale S Giovanni Bosco, Torino, site investigator) E Oddenino, MD (Department of Neurology, Ospedale Gradenigo, Torino, site investigator); C Geda, MD (Department of Neurology, Ospedale di Ivrea, and Department of Neurology, Ospedale di Chivasso, site investigator); C Doriguzzi Bozzo, MD (Department of Neurology, Ospedale di Pinerolo, site investigator), U Magliola, MD (Department of Neurology, Ospedale di Pinerolo, site investigator); D Papurello, MD (Department of Neurology, Ospedale di Ciriè, site investigator); P Santimaria, MD (Department of Neurology, Ospedale di Vercelli, site investigator); U Massazza, MD (Department of Neurology, Ospedale di Biella, site investigator); A Villani, MD (Department of Neurology, Ospedale di Domodossola, Advisory Committee) R Conti, MD (Department of Neurology, Ospedale di Domodossola, site investigator); F Pisano, MD (Fondazione Salvatore Maugeri, Clinica del Lavoro e della Riabilitazione, IRCCS, Scientific Institute of Veruno, site investigator); M Palermo, MD (Department of Neurology, Azienda Ospedaliera Santi Antonio e Biagio, Alessandria, site investigator); F Vergnano, MD (Department of Neurology, Ospedale di Casale Monferrato, site investigator); MT Penza, MD (Department of Neurology, Ospedale di Tortona, site investigator); N Di Vito, MD (Department of Neurology, Ospedale di Asti, site investigator); M Aguggia, MD (Department of Neurology, Ospedale di Asti, site investigator); I Pastore, MD (Department of Neurology, Azienda Ospedaliera S Croce e Carle, Cuneo, site investigator); P Meineri, MD (Department of Neurology, Azienda Ospedaliera S Croce e Carle, Cuneo, Advisory Committee); P Ghiglione MD (Department of Neurology, Ospedale di Savigliano, site investigator); D Seliak, MD (Department of Neurology, Ospedale di Savigliano, site investigator); C Cavestro, MD (Department of Neurology, Ospedale di Alba, site investigator); G Astegiano, MD (Department of Neurology, Ospedale di Alba, site investigator); G Corso, MD (Department of Neurology, Ospedale Regionale di Aosta, site investigator); E Bottacchi, MD Department of Neurology, Ospedale Regionale di Aosta, Advisory Committee).
Linked article 241513.
Funding This paper was supported in part by grants from Regione Piemonte (Ricerca Finalizzata 2002, grant No 12944; Ricerca Scientifica Applicata 2004, grant No A317) and Compagnia di San Paolo (grant No 2003.0078).
Competing interests None.
Ethics approval This study was conducted with the approval of the Comitato Etico Ospedale Molinette, Torino, Italy.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary