Article Text

Statistics from Altmetric.com
A 39-year-old man of Tunisian origin was admitted to hospital with abdominal pain and postprandial vomiting. A neurologist’s opinion was requested because of his pelvic and shoulder pain and a markedly elevated serum creatine kinase of 2321 U/L (<140 U/L). He had a history of asthma, recurrent epistaxis and hepatitis A. There were no details about his childhood or about his parents. His sister was well and he had no children.
On examination, he had normal eye movements and normal facial and neck strength. There was striking hypertrophy of the thigh muscles (figure 1), without atrophy, myotonia, rippling or muscle mounding. His limb strength, coordination and sensation appeared normal, but he had difficulty climbing two steps at a time.

(A) Hypertrophy of the musculature of the thighs. (B) Hypertrophy of the paraspinal musculature, evident on CT abdomen.
Electromyography showed diffuse neurogenic and myopathic motor unit potentials (both proximal and distal) with limited signs of denervation. MR scan of the thigh muscles showed no signal change. CT scan of abdomen showed a slightly steatotic appearance of the liver, with clear paraspinal muscles hypertrophy (figure 1).
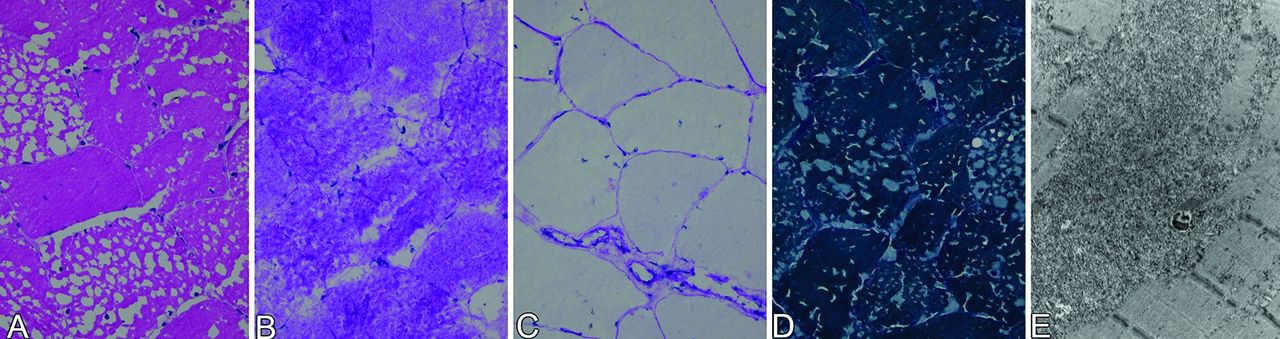
Light microscopy of a fresh-frozen right quadriceps muscle biopsy showed small vacuoles, reminiscent of ice crystal artefact. There was no inflammatory infiltrate. Gomori trichrome staining showed intracellular accumulation of an amorphous, blue–greyish substance, corresponding to periodic acid–Schiff-positive material, that was diastase sensitive. There was no lipid accumulation. However, electron microscopy showed a massive amount of non-membrane-bound glycogen, directly pointing to a glycogen storage disease. (figure 2).

{kind=link}
{kind=link}
(A) H&E stain showing vacuolisation of muscle fibres, reminiscent of ice crystal artefact. (B) Presence of periodic acid–Schiff-positive material in the vacuoles that were sensitive to diastase pretreatment (C). (D) Gomori trichrome stain showing intracellular accumulation of amorphic, blue–greyish substance. (E) Electron microscopy showing a massive amount of non-membrane-bound glycogen particles.
An ischaemic forearm test showed a flat lactate curve, with a normal rise of ammonia.
We diagnosed glycogen storage disease type III (debrancher enzyme deficiency). A red blood cell enzyme assay showed no enzyme activity at all, and the diagnosis was later confirmed by the genetic test showing homozygosity for the p.(Glu1072fs), c.3216_3217del mutation in exon 25 of the amylo-1,6-glucosidase (AGL) gene, a mutation previously reported in affected people with GSD III.
Glycogen storage disease type III is an autosomal recessive disease with a variable clinical picture. In childhood, there is typically hepatomegaly, with growth retardation and fasting hypoglycaemia (our patient had no childhood medical problems); later in life, some people develop a myopathy, often more distal than proximal. There may be wasting of leg muscles and intrinsic hand muscles, sometimes leading to an erroneous diagnosis of motor neurone disease or peripheral neuropathy.1
Our patient had a remarkable hypertrophy of the thigh and paraspinal muscles, which is not a usual characteristic feature of glycogen storage disease type III. Nevertheless, Marbini et al 2 described two brothers with this condition, observing pseudohypertrophy of sternocleidomastoid, trapezius and quadriceps muscles, together with mild distal wasting, suggesting that this type of pseudohypertrophy may be a distinctive feature of glycogen storage disease type III.2
The main learning point of this case is the value of muscle biopsy in patients with unexplained muscle enlargement; without it, this patient—with pseudohypertrophy, muscle weakness and raised serum creatine kinase—might have been assumed to have a muscular dystrophy, with extensive and futile genetic testing for a dystrophinopathy or limb girdle muscle dystrophy.
Footnotes
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement This paper was reviewed by Jon Walters, Swansea, UK.
Other content recommended for you
- Different clinical aspects of debrancher deficiency myopathy
- Muscle hypertrophy and pseudohypertrophy
- Metabolic myopathies: a practical approach
- The diagnostic value of hyperammonaemia induced by the non-ischaemic forearm exercise test
- McArdle disease: a clinical review
- The investigation and management of metabolic myopathies
- Proximal muscle weakness
- A case of Costello syndrome and glycogen storage disease type III
- Muscle diseases: mimics and chameleons
- Polymorphic markers of the glycogen debranching enzyme gene allowing linkage analysis in families with glycogen storage disease type III.