Article Text

Statistics from Altmetric.com
The history
A 45-year-old woman was brought to the emergency department by the police. She had been stopped for driving erratically and noted to be confused; an alcohol breathalyser test was negative. In the emergency department, she was disoriented in time and place, unable to give a history, but denied any symptoms.
The patient's sister had noted a behavioural change about 12 months previously when the patient began using cannabis, but otherwise she had functioned normally until 4 weeks before this presentation. Then her memory had deteriorated, her behaviour changed further and she began to have difficulties with routine tasks at work (eg, she required help to use a photocopier). She had been able to continue in her job as a lecturer, but colleagues had noticed her poor performance.
Two weeks before this admission, a neighbour found her in a confused state on the floor, having vomited and been incontinent of urine. After this, she complained of leg weakness which resolved. She did not seek medical attention at the time.
She had no medical or psychiatric history, and took no regular medication. She lived alone and worked as a lecturer in nursing. She drank 14 units of alcohol per week and had not smoked for 20 years. She had smoked cannabis for about 12 months but there was no other known recreational drug use. She had no travel history and no family history of neurological illness.
Examination
She was afebrile with no meningism. General medical examination was normal.
Although alert, she was disoriented in time and place and was unable to name the monarch, prime minister or dates of the Second World War. She scored 26/100 on the Addenbrooke's Cognitive Examination with marked deficits in all domains. She had difficulty following two-step commands. Cranial nerve examination was normal. She had no weakness, but the reflexes were brisk with upgoing plantars. There were no extrapyramidal, sensory or cerebellar signs.
Investigations
■ Blood and cerebrospinal fluid (CSF) results are summarised in box 1
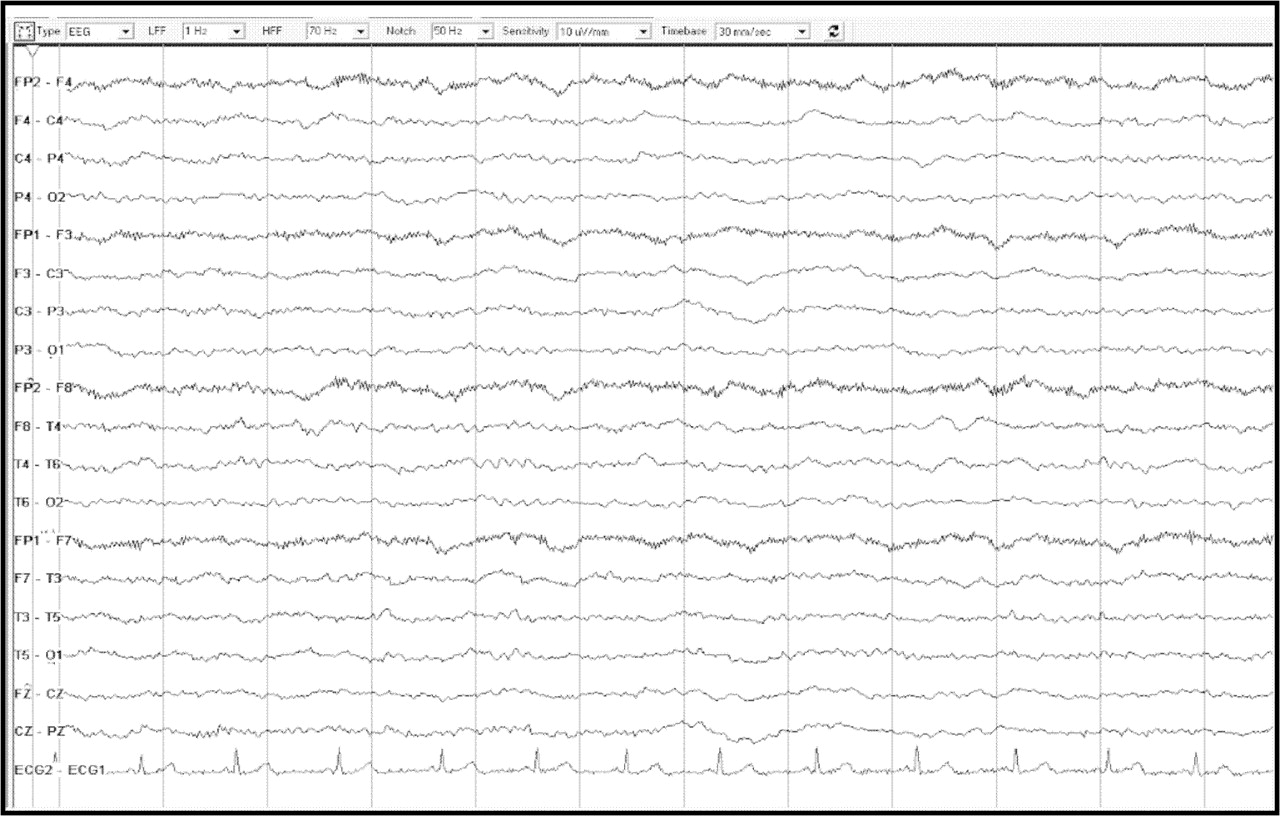
■ EEG
■ 18 March 2008: diffuse excess of slow activity consistent with an encephalopathic process, but no focal or epileptiform features (figure 1)
■ 26 March 2008: unchanged from 18 March 2008
■ Chest x-ray: normal
■ CT chest, abdomen, pelvis: normal
■ MRI head (18 March 2008): diffusely abnormal supratentorial white matter high signal without associated mass effect, with patchy high signal in the corpus callosum. Normal medial temporal structures (figure 2)
■ MRI head+MR venogram (28 March 2008): unchanged from 18 March 2008. No sinus or cortical vein thrombosis
■ MRI head 8 April 2008: improved but persistent supratentorial white matter changes. Increasing cerebral atrophy

EEG showing a diffuse excess of slow activity consistent with an encephalopathic process, but without focal or epileptiform features.

MRI reported as showing diffusely abnormal supratentorial white matter high signal without associated mass effect, with patchy high signal in the corpus callosum. Normal medial temporal structures.
Box 1 Investigations
Blood tests
■ The following were normal or negative:
■ FBC, B12, folate, ESR, U&Es, Ca, LFT, TFT, CRP, ACE, ammonia, lactate, blood lead, short Synacthen test, ANA, ENA, ANCA (PR3 and MPO), anti-TPO antibody, antineuronal antibodies, CMV IgG, Toxoplasma gondii IgG, hepatitis B surface antigen, hepatitis C antibody, HIV antigen/antibody, anticardiolipin IgG, white cell B galactosidase, MTTL1 (MELAS) genetic analysis
■ The following were abnormal or positive:
■ Measles IgG: test value 3.46 relative light units (positive>0.69)
■ Anticardiolipin IgM 12.5 MPL (reference 0–9.8); repeat sample 26.6 MPL (RVV time ratio and KC time ratio normal)
■ White cell arylsuphatase A 0.1 nmol/min/mgP (reference 0.4–1.6)
CSF examination
■ Opening pressure 17 cm CSF
■ WCC 0, RBC 30
■ CSF protein: 0.53g/l (0.14–0.45)
■ CSF glucose 3.2 mmol/l (serum 4.3)
■ Oligoclonal bands: negative
■ PCR for HSV1 and HSV2: negative
■ Intrathecal anti JC virus antibody: negative
■ 14-3-3 protein: positive
ANA, antineutrophil antibody; ANCA, antineutrophil cytoplasmic antibody; CMV, cytomegalovirus; CRP, C reactive protein; CSF, cerebrospinal fluid; ENA, extractable nuclear antigen; ESR, erythrocyte sedimentation rate; FBC, full blood count; HSV, herpes simplex virus; KC, kaolin clotting; LFT, liver function test; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; MPO, myeloperoxidase; PR3, proteinase 3; RBC, red blood cell; RVV, Russell viper venom; TFT, thyroid function test; TPO, thyroid peroxidase; WCC, white cell count.
Clinical progress
At the time of admission she was started on treatment, but her cognitive function deteriorated further. Three weeks after admission she began improving and subsequently recovered well. A further treatment was initiated, and she was transferred for rehabilitation. She was discharged home in June 2008 after 3 months in hospital.
She was reviewed as an outpatient 1 week after discharge. Neuropsychological assessment recorded only mild difficulties producing words beginning with a given letter and rather hesitant planning; otherwise, there were no abnormalities on tests or in general behaviour and style of performance. She returned to work 6 weeks later.
At a further outpatient review in November 2008, she remained well and was still working. She appeared cognitively normal, and tests of orientation, serial sevens, registration and recall were normal.
In December 2008, 9 months after her initial presentation, she was found dead at home. A postmortem was undertaken.
Dr Christopher Allen's discussion
When I started thinking about this case, the words of Geoffrey Donnan (from his clinicopathological conference (CPC)discussion here) came to mind: ‘One must consider two important principles: first, the CPC is the domain of the rare—either rare presentations of common conditions or common presentations of rare conditions—and second, that treatable conditions are rarely presented’. CPCs tend to have several other common characteristics: there must be pathology to show at the end; they are generally ‘interesting’ (also known as rare) conditions; the history is usually challenging and does not give the game away; and there are usually some clinical tests to show (such as CSF and neuroimaging). I also did a little background research, which revealed that in 60 previously published CPCs the most common diagnoses were: lymphoma (4), acute disseminated encephalomyelitis (3), subacute sclerosing panencephalitis (SSPE) (3) and central nervous system (CNS) angiitis (3).
So with this in mind let us review this case. The history is of 45-year-old female nurse lecturer, living alone, with no previous medical or psychiatric history and taking no medication. She was stopped for driving erratically and noted to be confused (the breathalyser test was negative). We are told she was disorientated in time and place, unable to give a history, but denied any symptoms. Her sister reported 12 months of behavioural change during which time she started using cannabis, but other than this she was normal until 4 weeks ago. Her deterioration can therefore be considered a subacute or acute encephalopathy (or ‘delirium’).
The situation is, however, complicated by the apparent preceding behavioural change, and several questions immediately come to mind. Most importantly, was this change neurological or psychological in nature? If neurological, it may be part of the same condition as the encephalopathy and would indicate more longstanding orbitofrontal or limbic dysfunction. On the other hand, it may have had a psychological basis—we are not told about other life events and stressors that may be relevant. If the latter is the case, some of the behaviours may have been contributory to the eventual presentation. For example, we know about the cannabis use, but this gives rise to the possibility of other illicit drug use (such as cocaine). And if other aspects of her behaviour, such as her sexual behaviour, also changed, sexually transmitted diseases such as HIV and syphilis come into the frame.
Coming to the acute presentation, she has a precipitate decline over 4 weeks with worsening memory and dyspraxia. We are told about an episode of self-resolving confusion with vomiting, incontinence and leg weakness. This may have been a seizure, but she did not seek medical attention and we do not know much more about this. There are no clues as to precipitants, either for her overall deterioration or for this episode. We are told she was a modest drinker and a smoker; cannabis is the only known illicit drug she was using (although she may have been taking others); and we are not told of any relevant family history or foreign travel. Unfortunately, there are few additional clues from the examination; we are told that ‘there was no fever, no meningism and general examination was normal’. This makes acute infective meningitis or encephalitis unlikely, and (although it is not explicitly stated) we can probably infer that there was no rash, no obvious internal abnormality such as organomegaly or heart murmurs, and there was nothing to suggest a ‘smelly’ inherited metabolic disease such as maple syrup urine disease or carnitine deficiency. Neurological examination revealed disorientation with a global cognitive failure, with the presentation more in keeping with encephalopathy than dementia. Although there was no weakness, there were some long tract signs in the form of brisk reflexes and extensor plantars, suggesting either brain pyramidal tract or spinal cord involvement. Otherwise there were no abnormalities in eye movements, and no cerebellar, extrapyramidal or sensory features.
So what is the ‘bedside syndrome’? This is a case of subacute encephalopathy, with potentially relevant distinguishing features being: a possible 1-year history of preceding fronto-limbic dysfunction; new onset of cannabis use; at least one recent self-limiting episode of altered awareness (possibly representing a seizure, drug toxicity or metabolic dysfunction); and pyramidal signs but no other focal neurological features.
The ‘bedside differential diagnosis’ here is wide and includes:
■ genetic causes (eg, mitochondrial disease, adult onset leukoencephalopathies, inborn error of intermediate metabolism);
■ vascular causes (eg, vasculitis, venous sinus thrombosis, antiphospholipid syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), amyloid angiopathy, multiple emboli);
■ drugs (eg, cannabis, cocaine, inhaled heroin, other self-poisoning);
■ infections (eg, HIV, syphilis, SSPE, Lyme disease, Whipple's);
■ autoimmune or inflammatory causes (eg, multiple sclerosis, acute disseminated encephalomyelitis, limbic encephalitis, Hashimoto's encephalopathy)
■ neoplastic/paraneoplastic causes (eg, multiple metastases, primary tumour, limbic encephalitis);
■ metabolic causes (eg, postanoxic, B12 deficiency, thiamine deficiency, porphyria);
■ degenerative causes (eg, variant Creutzfeldt–Jakob disease, fronto-temporal dementia).
So what do we learn from the investigations that may help us? The MRI is interesting. We are given the report of ‘diffusely abnormal supratentorial white matter high signal without associated mass effect, with patchy high signal in the corpus callosum. Normal medial temporal structures’ and two illustrative slices (figure 2). However, I have the benefit of having reviewed the complete MRI images, which clearly also show bilateral globus pallidus cavitation (figure 3). So this enables us to generate a rather more precise ‘clinicoradiological syndrome’ of subacute leukoencephalopathy with globus pallidus cavitation. Before refining our differential diagnosis, we should therefore review the causes of leukoencepahlopathy in adults, and then causes of globus pallidus cavitation.
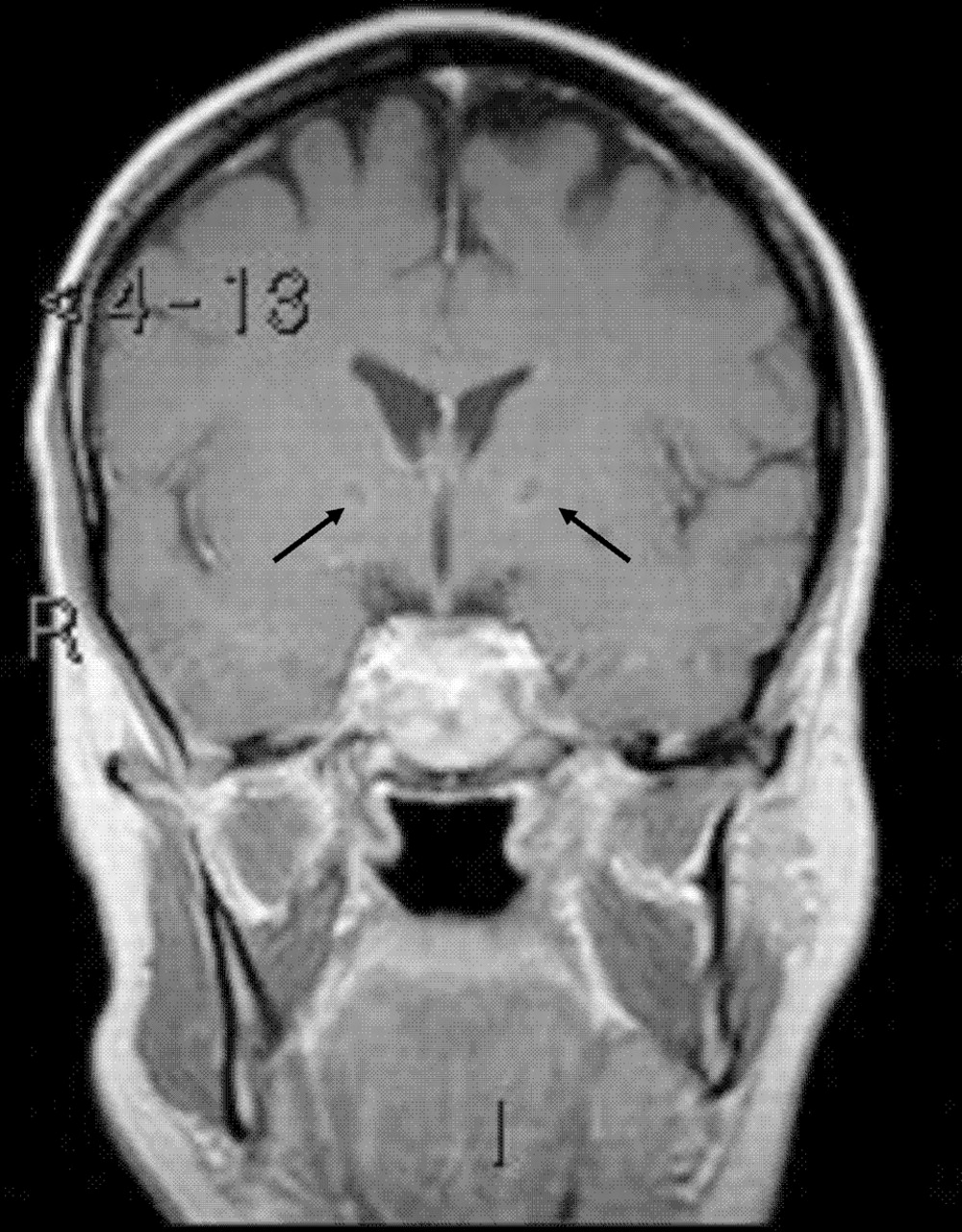
Additional MRI images showing bilateral globus pallidus cavitation (arrowed).
There are many causes of leukoencephalopathy in adults. Genetic causes can be divided broadly into two groups, complex molecular defects and defects in intermediary metabolism. Of the complex molecule defects, metachromatic leukodystrophy and adrenoleukodystrophy are the most well recognised, but the list of described conditions is long (box 2). And the most frequently encountered defects in intermediary metabolism are the mitochondrial respiratory chain disorders (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), etc) and the homocysteine remethylation defects (methylenetetrahydrofolate reductase (MTHFR) deficiency and cobalamin C disease), but again the complete list of conditions is more extensive (box 2). These are reviewed in detail in an informative review article by Sedel et al.1
Box 2 Genetic causes of leukoencephalopathy in adults
Complex molecule defects
■ Metachromatic leukodystrophy
■ Adrenoleukodystrophy
■ Krabbe's disease (globoid leucodystrophy)
■ Pelizaeus–Merzbacher disease
■ Fabry's disease
■ Cerebrotendinous xanthomatosis
■ Sjögren–Larsson disease
■ Vanishing white matter-like (adult)
■ Hereditary diffuse leukodystrophy with spheroids
■ Pigmentary orthochromic leukodystrophy
■ Austin's disease (multiple suphatide deficiency)
■ Canavan's disease
Defects in intermediary metabolism
■ Mitochondrial respiratory chain disorders (MELAS, etc)
■ Homocysteine remethylation defects (MTHFR deficiency and cobalamin C disease)
■ Amino acidurias (phenylketonuria, maple syrup disease)
■ Organic acidurias (eg, glutaric aciduria type 1)
MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; MTHFR, methylenetetrahydofolate reductase.
Toxic causes are the next big group of conditions to consider, with therapeutic drugs, environmental agents and drugs of abuse all potentially implicated. In this scenario, postanoxic and carbon monoxide (CO) poisoning are possible environmental agents that warrant consideration, and with the history of recent cannabis use, drugs of abuse are also possible causes for her presentation. Cocaine, ecstasy and inhaled heroin have all been associated with leukoencephalopathy, although there is no definite link with cannabis.
In terms of autoimmune and inflammatory conditions, limbic encephalitis (either autoimmune or paraneoplastic) would be a potential cause for a presentation of this type, as would a vasculitis affecting the CNS. Other inflammatory conditions including multiple sclerosis, acute disseminated encephalomyelitis and acute haemorrhagic encephalomyelitis, while possible, seem much less likely, and the pattern does not really point towards sarcoid or systemic lupus erythematosus although these conditions can present in a variety of forms and so should not be entirely dismissed.
A variety of chronic infections including HIV, progressive multifocal leukoencephalopathy, SSPE, syphilis and Lyme can present with a leukoencephalopathy. Primary CNS lymphoma and paraneoplastic syndromes such as limbic encephalitis are worthy of consideration, as is gliomatosis cerebri, although the imaging is not typical for this. Metabolic causes, particularly hypoxia, are in the differential; eclampsia, hypertensive encephalopathy and cobalamin or folate deficiency could cause some of these features but do not fit so well with the overall pattern. And vascular causes seem less likely, although CADASIL should be considered, and both Binswanger's disease and amyloid angiopathy can cause a leukoencephalopathy.
So what about the globus pallidus lesions? There are a number of recognised causes, which are summarised below (box 3). Several of these are clear possibilities in this case. Of the common causes, hypoxic ischaemic injury and CO poisoning are plausible; of the less common causes, drug abuse and Leigh syndrome should both be kept in the differential. And of the rare causes, cobalamin C disease, Wilson's disease and neurosyphillis should all be considered. The other causes listed would be very unlikely in the clinical context.
Box 3 Causes of globus pallidus lesions on MRI
Common
■ Hypoxic–ischaemic encephalopathy
■ Carbon monoxide poisoning
■ Neurofibromatosis type 1
Less common
■ Drug abuse
■ Hyperalimentation
■ Hepatic encephalopathy
■ Leigh's syndrome
■ Cyanide poisoning
■ Kernicterus
■ Hypothyroidism
■ Fahr's disease
Rare but important
■ Neurodegeneration with brain iron accumulation
■ Pantothenate kinase-associated neurodegeneration (formerly Hallervorden–Spatz disease)
■ Maple syrup urine disease
■ Methylmalonic acidaemia (cobalamin C disease)
■ Wilson's disease
■ Neurosyphilis (general paresis of the insane)
So, having identified the clinicoradiological syndrome and considered the causes of leukoencephalopathy and globus pallidus cavitation can we make a differential diagnosis? At this point, my list would be as follows:
■ Genetic
■ Leucodystrophy (metachromatic leukodystrophy, adrenoleukodystrophy)
■ Mitochondrial disease
■ Inborn errors of metabolism (homocysteine remethylation defects, eg, cobalamin C disease)
■ Wilson's disease
■ Toxic
■ Anoxia
■ CO
■ Cocaine, 3-4 methylenedioxymethamphetamine (ecstasy)
■ Heroin (inhaled)
■ Cannabis
■ Infection
■ HIV
■ Progressive multifocal leukoencephalopathy
■ SSPE
■ Neurosyphilis
■ Neoplastic
■ Primary CNS lymphoma
■ Paraneoplastic
■ Autoimmune
■ Limbic encephalitis
■ Vasculitis
■ Vascular
■ CADASIL
But now we need to consider the other information we are given, in particular the blood tests. First, what do we learn from the normal blood tests? The normal full blood count, erythrocyte sedimentation rate, urea and electrolytes, calcium, liver function tests and C reactive protein suggest there is no systemic metabolic or inflammatory condition, and the normal B12 and folate (and absence of macrocytosis) exclude some, although not all, remethylation defects. A normal ACE would go against sarcoidosis, and normal ammonia would effectively exclude a urea cycle defect (such as ornithine transcarbamylase deficiency). The normal lactate makes MELAS less likely, and normal lead excludes lead poisoning (eg, from adulterated cannabis). We know she is not hypoadrenal, due to the normal short Synacthen test, which goes against adrenoleukodystrophy (manifesting carrier of this X linked disorder), and the normal autoantibodies make lupus, systemic vasculitis and paraneoplastic causes much less likely. The negative tests for HIV, hepatitis B and C and toxoplasma effectively exclude these conditions. Normal white cell β-galactosidase excludes GM1-gangliosidosis and a normal MTTL1 gene excludes 80% of MELAS cases.
So what about the abnormal blood tests? There are a few. First, the measles immunoglobulin G (IgG) titre is elevated; however, this merely reflects past exposure or vaccination and so can be dismissed as irrelevant. Second, anticardiolipin IgM is elevated on two separate tests, although Russell viper venom time ratio and kaolin clotting time ratio are normal. I think this is also a red herring: false positive anticardiolipin tests are not uncommon (biological false positives without prior treponemal infection). A diagnosis of antiphospholipid syndrome requires at least one of the clinical and one of the laboratory criteria listed below to be met (box 4)2; our patient does not meet any of these. So I think this finding is also irrelevant.
Box 4 Criteria for a diagnosis of antiphospholipid syndrome (at least one clinical and one laboratory criterion is required for the diagnosis)2
Clinical
■ A documented episode of arterial, venous, or small vessel thrombosis—other than superficial venous thrombosis—in any tissue or organ by objective validated criteria with no significant evidence of inflammation in the vessel wall
■ and/or
■ One or more unexplained deaths of a morphologically normal fetus (documented by ultrasound or direct examination of the fetus) at or beyond the 10th week of gestation and/or three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes excluded or at least one premature birth of a morphologically normal neonate before the 34th week of gestation due to eclampsia or severe pre-eclampsia according to standard definitions, or recognised features of placental insufficiency
■ plus one of
Laboratory
■ Anticardiolipin IgG and/or IgM measured by standardised, non-cofactor dependent ELISA on two or more occasions, not less than 12 weeks apart; medium or high titre (ie, >40 GPL or MPL, or >99th percentile) and/or
■ Anti-β2 glycoprotein I IgG and/or IgM measured by standardised ELISA on two or more occasions, not less than 12 weeks apart; medium or high titre (>99th percentile) and/or
■ Lupus anticoagulant detected on two occasions not less than 12 weeks apart according to the guidelines of the International Society of Thrombosis and Haemostasis
However, the white cell arylsuphatase A (ArSA) is clearly low at 0.1 nmol/min/mgP (reference range 0.4–1.6); this is 25% of the lower limit of normal. The question here is: is this low enough to make a diagnosis of metachromatic leukodystrophy? Or is this pseudodeficiency?
Data from the Willink lab in Manchester (where our samples from Cambridge are sent) suggest that most patients with metachromatic leukodystrophy have ArSA levels of 10% of the lower limit of normal (or lower). However, confirmed metachromatic leukodystrophy cases have been documented with ArSA levels at up to 20% of the lower limit of normal. Prior to performing genetic tests for metachromatic leukodystrophy, the lab recommends confirming low ArSA levels with a repeat sample; we just have the one result here. Set against this background, ArSA pseudodeficiency, in which enzyme activity is significantly reduced but not sufficiently to cause the metachromatic leukodystrophy state, is said to occur in 0.5–2% of the population. This seems more likely to be the situation in this case. Interestingly, ArSA pseudodeficiency (10–30% of normal enzyme activity) has been reported in association with posthypoxic demyelination.3
In terms of other tests there is little extra to be learnt. The EEG shows just generalised non-specific slowing, and the ECG trace (from the EEG) looks normal. The CSF is essentially normal, with no evidence of CNS inflammation; the only abnormality is a positive 14-3-3 protein, but this is a very non-specific feature.
It is also worth considering tests which were not done but which might have been helpful. There is quite a list, including:
■ a drug screen (for drugs other than cannabis);
■ peripheral neurophysiology (for metachromatic leukodystrophy, adrenoleukodystrophy, MTHFR deficiency, cobalamin C disease);
■ antivoltage-gated potassium channel and antiN-methyl-D-aspartate antibodies (for limbic encephalitis);
■ urinary suphatide/ArSA gene (for metachromatic leukodystrophy);
■ very long chain fatty acids (for adrenoleukodystrophy);
■ leucocyte galactocerebrosidase (for Krabbe's disease);
■ leucocyte α-galactosidase (for Fabry's disease);
■ serum homocysteine/methylmalonic acid (for cystathionine B-sulphase, MTHFR deficiency, cobalamin C disease);
■ plasma cholestanol (for cerebrotendinous xanthomatosis);
■ urinary organic acids (for methylmalonic aciduria and other inborn errors of metabolism);
■ CSF lactate and other mitochondrial genes (for mitochondrial disorders);
■ caeruloplasmin and urinary copper (you should never forget Wilson's!);
■ urinary porphobilinogen (for acute intermittent porphyria);
■ specific serology for syphilis and borrelia, CSF cytology (for lymphoma);
■ MRI cervical spine (for cobalamin C disease);
■ 12-lead ECG (for long QT syndrome).
So, we can refine the list of potential diagnoses in the light of these additional investigations. I think a toxic cause, particularly postanoxia, is a distinct possibility (maybe in the context of ArSA pseudodeficiency). Unrecognised cocaine use could cause this presentation, as could several genetic conditions including mitochondrial disease and cobalamin C disease. Neurosyphillis is possible, as is limbic encephalitis, but these are less likely in the overall clinical context, and the remainder of the differential diagnosis discussed above seems increasingly unlikely.
So before making a final diagnosis, are there any final clues from the clinical progression?
We are told that on admission she started a treatment, but initially deteriorated further. Was this treatment an anti-infective agent such as antibiotics or acyclovir, or was she given steroids at this point? We do not know. We do know that after 10 days she had a further MRI and MR venogram; however, it is not clear whether this was because the diagnosis was still unclear at that stage, or whether the diagnosis which had been reached raised the suspicion of a thrombotic tendency. We are also told that 3 weeks after the admission she began improving and that another treatment was initiated; however, it is not clear whether this treatment was started prior to any improvement or if it was initiated after a spontaneous improvement had already begun. The nature of this treatment is also unknown; it could be IVIg or plasma exchange for an autoimmune condition, anticoagulants for antiphospholipid syndrome, a treatment for drug abuse (including psychotherapy), treatment for an infection (eg, neurosyphilis), a specific treatment for MTHFR deficiency, cobalamin C disease or another inborn error of metabolism or even a treatment for Wilson's disease (you should never forget Wilson's!).
We are then told that an MRI head, repeated after 3 weeks, shows improvement but with some residual white matter changes and increasing cerebral atrophy. Was the clinical and radiological improvement because of, or despite, treatment? This sort of remitting pattern could fit with delayed postanoxia, but also with an inflammatory condition or a mitochondrial disorder or inborn error of metabolism. She was discharged home from rehabilitation after 3 months, by which time her cognition was good enough for her to return to work 2 months after discharge, and by 8 months she seems to have recovered completely. This would be very much against metachromatic leukodystrophy or other leukodystrophy, and would put us in the category of treatable or spontaneously relapsing and remitting conditions. Possibilities would include: mitochondrial disorders, MTHFR deficiency or cobalamin C disease, delayed postanoxic leukoencephalopathy, an inflammatory or infective disorder, a prothrombotic condition and, just possibly, Wilson's disease.
But then, despite her improvement, we are told that 9 months later she was found dead at home. This appears to be a sudden unexpected death, and so a variety of direct causes of death should be considered, including cardiovascular (pulmonary embolus, myocardial infarction), cerebrovascular (possibly haemorrhage from anticoagulants), a cardiac arrhythmia, suicide or drug overdose, seizure, or recurrence of a metabolic encephalopathy. So finally let us take a step back and reconsider the overall course (figure 4); the presentation is of a reversible leukoencephalopathy followed by sudden death.

Graphical representation of the disease time course.
Taking everything into consideration, my first bet would be cobalamin C disease with ArSA pseudodeficiency as a cause for the subacute encephalopathy, with death resulting from a massive pulmonary embolus (due to the associated hyperhomocysteinaemia). A series of patients with cobalamin C disease and with similarities to this case has been published.4
However, I would like to make a couple of ‘side bets’! I think this could also be a mitochondrial disease (more typical of Leigh disease than MELAS), exacerbated by cannabis smoking, and with death caused by a recurrence of encephalopathy. Alternatively, this could be delayed posthypoxic encephalopathy (after drug overdose or cardiac arrhythmia) in a patient with ArSA pseudodeficiency, with death caused by a further overdose or arrhythmia.
Pathology: Professor James Ironside
As we have heard, the patient was found dead in her flat. At that time, and at postmortem, she had ‘cherry red’ discolouration of the skin, and haematological analysis revealed raised levels of carboxyhaemoglobin.
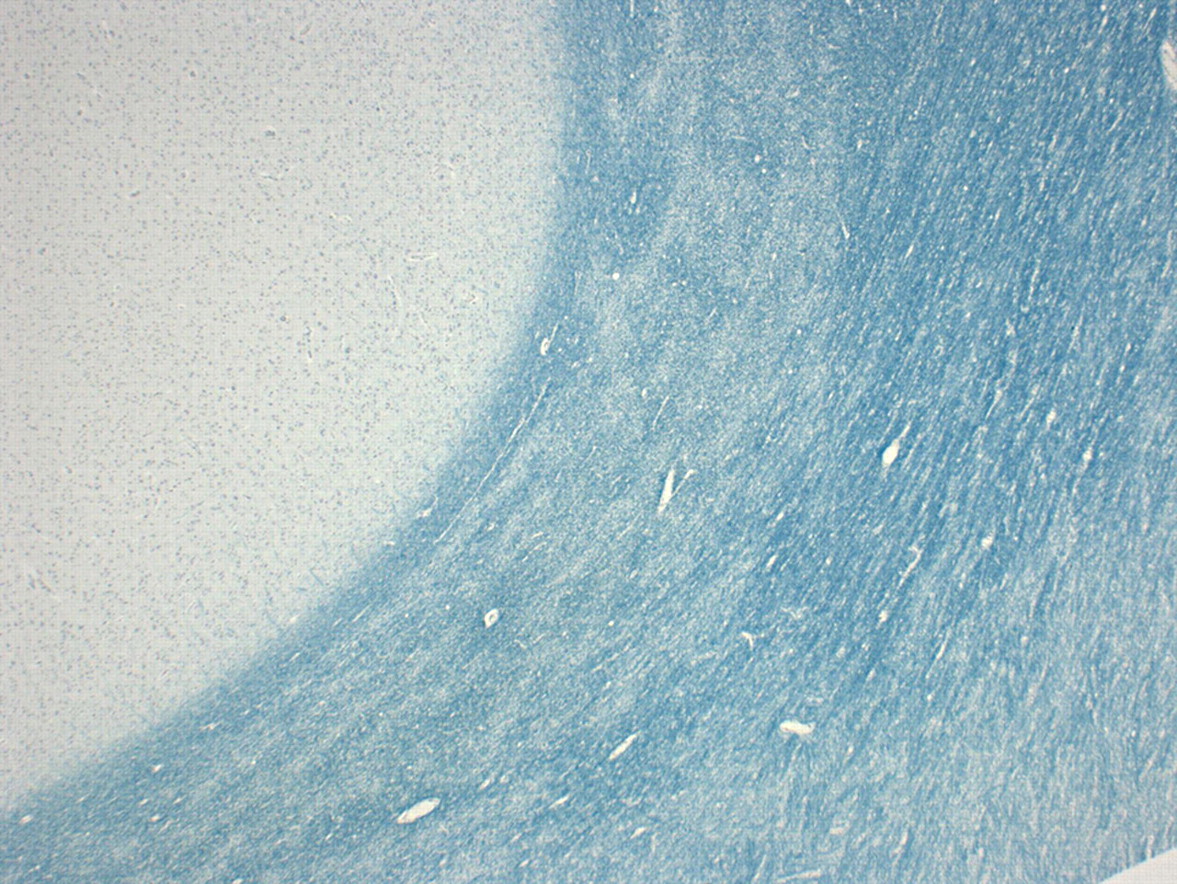
In terms of pathological specimens, the brain was not retained but samples were taken for histological assessment. The brain itself was of a pink appearance (figure 5); under low powered microscopy, there was clear putaminal cavitation and vascular mineralisation. Under higher power on H&E staining, there were Rosenthal fibres in the putamen; these can be a feature of chronic gliotic change, metabolic diseases (including Alexander's disease) and some tumours (particularly juvenile pilocytic astrocytomas). Elsewhere, there was evidence of white matter rarefaction and gliosis, with widespread myelin pallor on Luxol fast blue staining (figure 6). Taken together, these findings of putaminal necrosis, chronic gliosis and widespread myelin breakdown (known as ‘Grinker’s myelinopathy') are typical of CO toxicity and suggest chronic exposure.
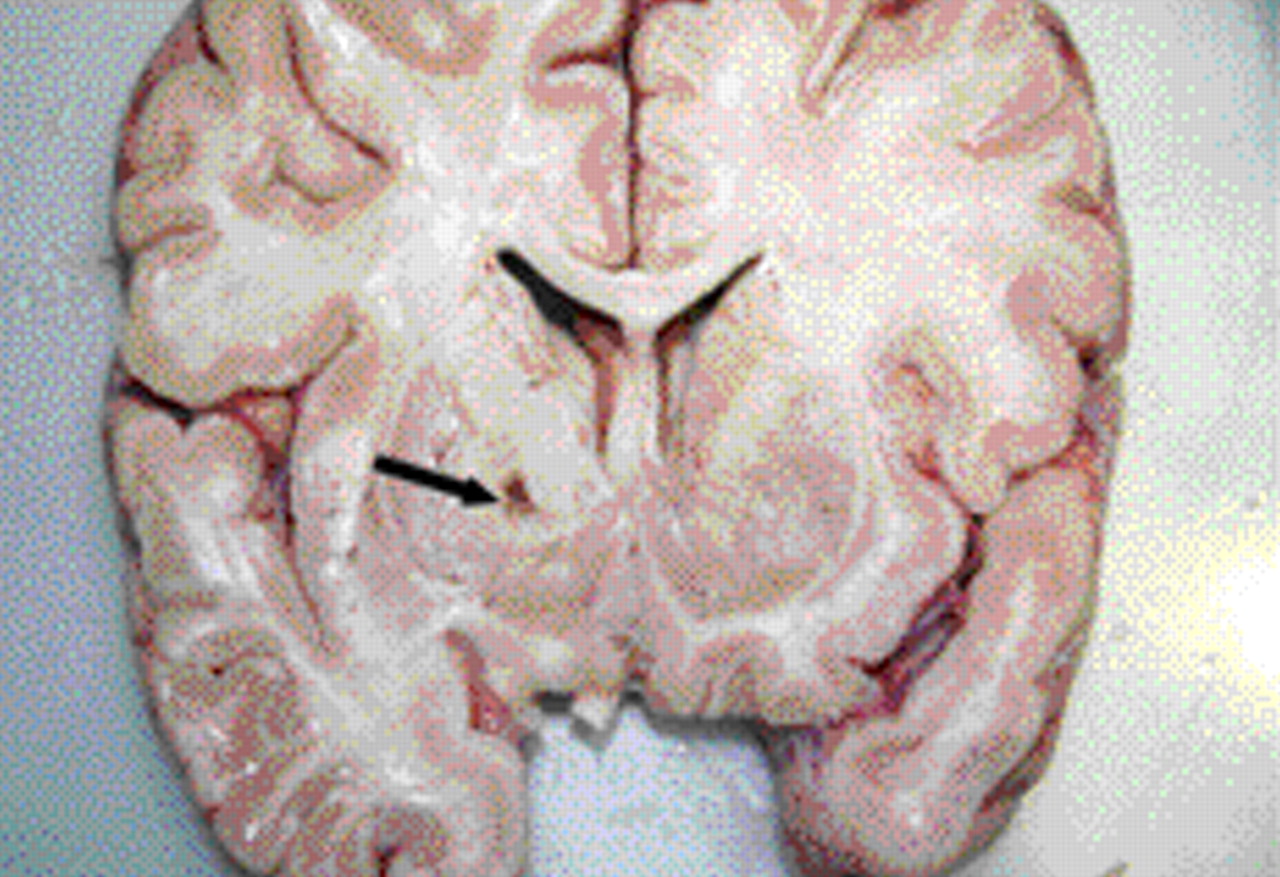
Pathological specimen of the patient's brain, with typical ‘pink brain’ appearance and putamen lesion (arrowed).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Low powered sections of brain stained with Luxol fast blue stain showing myelin pallor.
Discussion
Dr Jonathon Schott: Just a comment, particularly for any trainees here. In a presentation of this sort, voltage-gated potassium channel antibodies should always be checked. While this is not the diagnosis here, an autoimmune encephalopathy is a potentially treatable cause of subacute cognitive decline that should always be considered and excluded.
Audience member: Was carbon monoxide poisoning considered on the basis of the MRI findings?
Dr Richard Davenport: Toxic causes were raised as a possibility, but as Dr Allen has described, the differential diagnosis for this clinicoradiological pattern is extensive.
Dr Chris Allen: I had dismissed the possibility of carbon monoxide poisoning as she was found driving her car, rather than with a hosepipe into the car.
Comment
CO poisoning can occur from a variety of sources including gas heaters, automobile exhaust fumes and smoke; in this case, the patient was found to have a faulty gas heater in her home emitting high levels of CO.
CO crosses the alveolo-capillary membrane and binds to haemoglobin with an affinity 200 times that of oxygen. As a result it inhibits oxygen transfer, producing tissue hypoxia; it may also have more direct effects on brain oxidative metabolism.5 Clinically, CO may result in acute or chronic poisoning, with the severity of symptoms broadly related to the concentration of CO in the blood. The CNS and heart are most affected by CO.
Acute poisoning tends to present initially with headache and dizziness; as blood concentrations rise, impairments of higher cerebral function may appear, with nausea, vomiting and shortness of breath. At higher levels, cardiac arrhythmias, delirium, hallucinations, seizures and coma may develop; respiratory arrest and death may eventually supervene. It is worth noting that the ‘cherry red’ skin discolouration, associated with CO poisoning and noted at postmortem in this case, is usually only of use to the pathologist rather than the clinician or patient. The identification of this discolouration is unreliable in vivo6 and it should not be considered a useful discriminatory feature in the acute setting.
If acute or chronic CO poisoning is suspected, treatment is aimed at eliminating CO from the body as quickly as possible, and counteracting associated tissue hypoxia. Patients should be removed from the CO source and administered 100% oxygen as soon as possible. The diagnosis can be confirmed by measurement of the blood CO level. There is evidence that patients with impaired consciousness or blood CO levels of greater than 30% benefit from hyperbaric oxygen treatment. This treatment may also reduce the incidence of delayed neurological complications (discussed below).5
Chronic poisoning occurs in long-term exposure to low CO levels, and usually causes non-specific symptoms including headache, nausea, depression, poor concentration and confusion. It is not clear whether chronic exposure to low levels of CO cause permanent damage to the CNS, but symptoms usually resolve upon removal of the CO source.
The delayed sequelae of acute CO poisoning are neurological and psychiatric features that appear 1–3 weeks after an episode of acute CO poisoning, often after complete recovery from the initial episode. The incidence of these symptoms may be in the order of 15–40% of individuals undergoing acute CO poisoning.5 A wide variety of neurological features can present in this setting, but amnesia and other disorders of cognitive function, Parkinsonism, personality change and psychiatric conditions are well described. Other neurological features include multiple sclerosis-like symptoms, seizures, supranuclear gaze palsy, cerebral haemorrhage, peripheral neuropathy and cortical blindness.5 Non-neurological features may occur and include bullous skin lesions, cardiotoxicity and muscle necrosis with renal failure.7 Risk factors for delayed sequelae include older age and severity of initial anoxia.8 9 The long-term outcome is relatively positive, with 50–75% recovering completely.8
One specific delayed manifestation is delayed CO encephalopathy, which characteristically presents as a subacute encephalopathy several weeks after an episode of acute CO toxicity. The exact causative process is controversial, but postulated mechanisms are direct toxicity of CO, cerebral blood vessel damage, cerebral oedema and a hypersensitivity reaction; the fact that a similar process follows cerebral anoxia from other causes suggests tissue anoxia is important,5 although unlikely to be the only mechanism involved.10 The result is delayed reversible white matter demyelination, known as Grinker myelinopathy, which can progress to irreversible necrosis within the brain.5 In addition to Grinker myelinopathy, basal ganglia (particularly globus pallidus) degeneration and necrosis is well described; the histology of these lesions is similar to that seen in other anoxic conditions. Similar changes may occur in other grey matter structures including cerebral cortex and hippocampus.
The precise sequence of events in our patient was uncertain, but at least one episode (and possibly several) of acute CO intoxication appears to have precipitated delayed CO encephalopathy, prompting the initial admission. She made a good recovery, almost certainly unrelated to the treatments given (acyclovir and corticosteroids) and returned to work. This period of good health corresponds to the summer months, when presumably she did not need to use her gas fire. She then died suddenly in December, presumably due to acute CO poisoning, and subsequently the gas fire was identified as the source of the CO.
The significance of the ArSA pseudodeficiency is unclear, but conceivably this rendered her more susceptible than usual to CO toxicity. Postanoxic encephalopathy may result from other causes (eg, respiratory arrest)11 12 and may be associated with ArSA pseudodeficiency.3 One could speculate that ArSA pseudodeficiency predisposed to CO encephalopathy in this case.
While delayed sequelae of CO poisoning usually follows a recognised episode of acute CO toxicity, in some cases (like this one) the acute episode may not be recognised. Considering this condition in the differential may lead to timely diagnosis and enable removal of any source of CO. This is therefore an unusual CPC in that the condition was potentially treatable.
Dr Chris Allen's clinical diagnosis
■ Cobalamin C disease with ArSA pseudodeficiency with death from a pulmonary embolus
Alternative diagnoses of:
■ Mitochondrial disease (Leigh disease) exacerbated by cannabis smoking and with death caused by a recurrence of encephalopathy.
■ Delayed posthypoxic encephalopathy (after drug overdose or cardiac arrhythmia) in a patient with ArSA pseudodeficiency, with death from further overdose or arrhythmia
Professor James Ironside's pathological diagnosis
■ CO poisoning, with evidence of chronic exposure
Acknowledgments
We are very grateful to the patient's family for allowing us to discuss and publish this case.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed. Reviewed by Luke Bennetto from Bristol, UK.
Other content recommended for you
- Chronic and occult carbon monoxide poisoning: we don’t know what we’re missing
- Carbon monoxide poisoning
- Acute carbon monoxide intoxication during pregnancy. One case report and review of the literature
- Delayed postanoxic encephalopathy after carbon monoxide poisoning
- Carbon monoxide poisoning mimicking long-QT induced syncope
- Carbon monoxide poisoning: correlation of neurological findings between accident and emergency departments and a hyperbaric unit
- CO poisoning seems to spare memory
- Acute carbon monoxide poisoning as a cause of rhabdomyolysis in a case of flame burn
- Effects of Carbon Monoxide Poisoning on Neutrophil Responses in Patients Treated with Hyperbaric Oxygen
- Hyperbaric oxygen in carbon monoxide poisoning