Article Text

Abstract
Parkinson's disease (PD) is a common neurodegenerative condition that usually presents with symptoms related to asymmetric bradykinesia, resting tremor, rigidity and postural instability. Making the correct diagnosis can be challenging as many conditions—including tremor, gait and atypical parkinsonian disorders—can mimic PD. PD can present with unanticipated motor and non-motor symptoms, and so can masquerade as a number of rheumatological, neurological, sleep and mood disorders. Careful clinical assessment, informed by well-validated diagnostic criteria, is important in the initial diagnostic formulation. In uncertain or ambiguous cases, follow-up with monitoring of the treatment response usually gives the correct diagnosis, as validated in postmortem follow-up studies. ‘Premotor’ PD—a range of non-motor symptoms, particularly sleep disorders and constipation, which can occur up to 20 years before PD motor onset—is common. The presence of non-motor features in early disease sometimes supports the diagnosis of PD. Here we give an overview of the diagnosis of PD and its most important chameleons and mimics, and review the recent advances in structural and functional imaging in parkinsonism.
- PARKINSON-S DISEASE
- LEWY BODY
- DYSTONIA
- HUNTINGTON-S
- MULTISYSTEM ATROPHY
Statistics from Altmetric.com
Introduction
Parkinson's disease (PD) is a common clinical condition affecting approximately 1% of the population older than 65 years.1 ,2 Many lay people and non-specialists inevitably associate PD with tremor. However, tremor is a presenting feature in only half of all patients with PD, although 90%–100% of patients with PD have tremor at some stage in their disease course.3–6 There are other common causes of tremor—as well as conditions that cause slowness or a change in gait and posture—that can present as PD mimics.7 Our concept of PD has changed from a pure movement disorder to a clinicopathological entity with protean features, including changes in mood, sleep, behaviour, cognition and autonomic function. PD can cause symptoms that lead to referrals to psychiatry, general medicine, care of the elderly, orthopaedic and rheumatology clinics, before arriving at the correct primary underlying diagnosis. A definite diagnosis of PD can only be made at postmortem by identifying degeneration of the substantia nigra pars compacta and the presence of Lewy bodies—insoluble cytoplasmic inclusions containing aggregated alpha-synuclein. Although early diagnosis is occasionally difficult, by the time of death at least 90% of postmortem-proven patients with PD have a correct clinical diagnosis.8
Typical PD and the clinical examination
The distinctive pathological features of PD have led to robust and well-validated clinical diagnostic criteria. In 1988, Gibb and Lees developed the Queen Square Brain Bank (QSBB) criteria for PD, based on careful clinical observation combined with clinicopathological correlation.9 In 1992, Hughes and colleagues showed that only 76% of patients diagnosed as having PD by UK neurologists fulfilled the neuropathological diagnostic criteria for the disease. Retrospective application of the QSBB clinical criteria improved the diagnostic accuracy to 82%. Multiple system atrophy, progressive supranuclear palsy, Alzheimer's disease, Alzheimer-like pathology and vascular parkinsonism comprised most of the alternative diagnoses.10 The same group's subsequent study in 2001 showed that the clinical diagnosis accuracy had improved to 90%.11 The QSBB criteria remain a benchmark for PD diagnosis, and careful application and appreciation of the criteria will prevent most pitfalls for the clinician. However, it is important to appreciate that many patients with PD may not fulfil the QSBB criteria for PD at presentation (box 1). Expert clinicians are reportedly more accurate than the QSBB criteria at diagnosing PD, and this presumably relates to the integration of a number of sometimes subtle historical and examination findings.8 ,11 Parkinson's syndrome (stage 1 of the QSBB criteria) is defined by bradykinesia and one additional feature of tremor, rigidity or postural instability. Bradykinesia can be appreciated as a general slowness and paucity of movement, particularly in a decrease in the normal spontaneous fidgeting and gestural movements that occur during the interview. Parkinsonian bradykinesia is specifically elicited by examining repeated movements, usually finger taps, hand grips, pronation–supination movements, toe taps and heel stamps, following the scheme of the Movement Disorders Society unified PD rating scale (MDS-UPDRS). In PD, there is a specific form of bradykinesia involving progressive loss of the speed and amplitude of repetitive movements and there may be arrests in movements and ‘re-setting’. There is also a perceptible slowness of initiation and subtle weakness. The MDS-UPDRS scheme mandates that these movements are assessed over 10 repetitions.12 Many other factors can affect the ability to carry out repeated movements, including arthritis, pyramidal deficits, dyspraxia, depression, obsessional slowness, cerebellar disease, dystonia and other cognitive deficits. These concurrent comorbidities may make identification of true bradykinesia difficult in some patients, particularly the elderly. It is important that parkinsonian bradykinesia is not ‘over-diagnosed’ and these PD–bradykinesia mimics are a potential pitfall for the unwary. Bradykinesia can be difficult to assess in patients with marked postural tremor. If there is uncertainty as to whether there is genuine parkinsonian bradykinesia, we refer to the abnormality of movement as ‘clumsiness’ or of ‘slowness of movement without decrement’, pending further diagnostic information. A history of fatigable movement (‘I can start brushing my teeth normally but then the movements fade out’), the presence of generalised bradykinesia and the ability to assess multiple movements (ie, arms and legs) as outlined above can help in assessing ambiguous cases.
UK Queen Square Brain Bank Criteria for the diagnosis of Parkinson's disease9
Step 1 Diagnosis of a parkinsonian syndrome
Bradykinesia (slow to initiate voluntary movement with progressively reduced speed and amplitude of repetitive actions) and at least one of the following:
-
Muscular rigidity
-
Rest tremor (4–6 Hz)
-
Postural instability unrelated to primary visual, cerebellar, vestibular or proprioceptive dysfunction
Step 2 Exclusion criteria of Parkinson's disease
History of:
-
Repeated strokes with stepwise progression
-
Repeated head injury
-
Antipsychotics or dopamine-depleting drugs
-
Definite encephalitis and/or oculogyric crisis on no drug treatment
-
More than one affected relative (Note: this exclusion criteria is usually ignored)
-
Sustained remission
-
Negative response to large doses of levodopa (if malabsorption excluded)
-
Strictly unilateral features after 3 years
-
Other neurological features: supranuclear gaze palsy, cerebellar signs, early severe autonomic involvement, Babinski's sign, early severe dementia with disturbance of language, memory or praxis
-
Exposure to known neurotoxin
-
Presence of cerebral tumour or communicating hydrocephalus on neuroimaging
Step 3 Supportive criteria for Parkinson's disease
Three or more required for the diagnosis of definite Parkinson's disease
-
Unilateral onset
-
Rest tremor present
-
Progressive disorder
-
Persistent asymmetry affecting the side of onset most
-
Excellent response to levodopa (70%–100%)
-
Severe levodopa-induced chorea
-
Levodopa response for more than 5 years
-
Clinical course of over 10 years
-
Box courtesy of Professor Andrew Lees
It is important to appreciate that fulfilling stage 1 criteria is not equivalent to fulfilling the QSBB diagnostic criteria for PD as there should be no exclusion criteria, and three supportive criteria should be present (box 1). The supportive criteria include the presence of a typical 4–6 Hz resting tremor. Patients without a typical resting tremor are unlikely to fulfil the QSBB criteria for PD at presentation, as six of the other features relate to disease course and treatment response. Resting tremor can be assessed in clinic by asking the patient to relax in a chair with arms supported and asking them to carry out a distraction task, such as counting backwards or lying on the couch in a relaxed position. In PD, the tremor normally stops on starting a movement but may re-emerge with sustained posture (re-emergent tremor). PD tremor can affect the lips, chin and legs, while head and neck tremor points towards other disorders.3 PD is an asymmetrical disease, and asymmetry at onset and through the disease course are both important supportive criteria. Additional features that have emerged since the publication of the QSBB criteria—and that may help to support a diagnosis of PD early in the disease course—include anosmia, prodromal constipation and rapid eye movement (REM) sleep behaviour disorder. Observing the clinical course and progression of a patient with suspected PD is one of the most important aspects of making an accurate diagnosis.13 PD is progressive, and disability related to cognition and gait usually occurs more than 5 years after disease onset. The response to drug treatment is an important part of the assessment. Patients with PD usually respond well to levodopa, but 20%–40% of patients develop levodopa-induced dyskinesia within 5 years, although in most patients this is not disabling.14 In PD, particularly in patients with akinetic–rigid dominant presentations, there is usually a marked (>70%) beneficial response to levodopa treatment.9
PD chameleons
Failure to recognise PD can relate to non-neurological or atypical presentations of motor and/or non-motor symptoms (box 2). PD causes stiffness and slowness, which also occur in a plethora of rheumatological conditions, leading to missed and delayed diagnoses. Many patients also have a prodromal non-motor syndrome related to underlying Lewy body pathology. Identifying patients with prodromal PD or ‘Parkinson's at risk’ subjects is now a major research endeavour,15 with a view to starting early disease-modifying therapy. From a clinical standpoint, we recommend regular follow-up of patients with, for example, REM sleep behaviour disorder, as they have an increased risk of developing PD, multiple system atrophy and dementia with Lewy bodies.16–18
Parkinson's disease chameleons
Non-motor related
Sensory features and pain
Sleep-related disorders
Autonomic features (urinary urgency)
Mood disturbance (depression and anxiety)
Motor related
Stroke
Spinal degenerative disease
Frozen shoulder
Asymmetric motor performance, for example, difficulty in swimming in a straight line or skiing
Exercise-induced dystonia
Note: Bold entries are common presentations
These motor and non-motor symptoms can lead to referral to various specialties, including orthopaedics, spinal surgery, urology, psychiatry, care of the elderly and general practice.16 PD can also be misdiagnosed as any of its mimics mentioned below.16
Non-motor symptoms
It is well established that patients with PD can have non-motor symptoms before developing the motor features of PD. One study showed that 21% of autopsy-proven patients with PD presented initially with exclusively non-motor symptoms.8 Patients who presented with non-motor symptoms had a delayed diagnosis of PD at 1.6 years compared with 1.0 years for patients with motor presentations.8
Sensory symptoms: Pain is the most frequent non-motor presentation of PD, accounting for up to 15% of all PD presentations and is, therefore, the most common PD chameleon.8 Pain in PD most likely relates to the motor-affected side. Many patients with PD have been previously diagnosed to have osteoarthritis, degenerative spinal disease and frozen shoulder due to both non-motor (pain) and motor symptoms. Some patients may be referred to orthopaedic surgeons, spinal surgeons or rheumatologists and may have unnecessary procedures,8 ,16 although some patients also develop musculoskeletal disorders secondary to parkinsonian bradykinesia and stiffness. Hyposmia is a common non-motor symptom in PD, occurring in 80%–90% of patients and preceding the motor symptoms by up to 40 years. In a large prospective study, impaired olfaction predated the diagnosis of PD by 4 years.19 However, it is rarely an isolated presenting symptom.17 ,18 Hyposmia can occur in asymptomatic first-degree relatives of patients with familial PD.20
REM sleep behaviour disorder is usually reported by the patient's partner and can precede the diagnosis of PD by many years. The witness may report vivid dreams of a violent nature with the patient screaming, yelling and punching during sleep.21 Several studies report that 80% of patients with the idiopathic form who present to sleep disorders clinics eventually develop a neurodegenerative disorder—most commonly PD—but sometimes include other synuclein disorders such as multiple system atrophy and dementia with Lewy bodies.22–24 However, REM sleep behaviour disorder is rare at the presentation of PD, with one study showing a prevalence of <4% at initial assessment. It is particularly associated with visuospatial and cognitive dysfunction; the development of REM sleep behaviour disorder predicts the development of daytime hallucinations.8 ,25 ,26
Autonomic features: Autonomic features are common in PD; some, particularly constipation, can precede the movement disorder. Patients may report increased difficulty passing faeces and incomplete bowel emptying; this may prompt multiple gastroenterological investigations before the underlying diagnosis of PD becomes apparent. Constipation can predate the motor features of PD by up to 12 years.27 A small proportion (4%) of all patients with PD present with urinary urgency.8 This can lead to misdiagnosis of benign prostatic hypertrophy.16 In one large prospective study of early PD, hypersalivation and dribbling occurred in 55% of patients and urinary urgency was reported in 46% of patients within the first year of diagnosis.28 In another prospective study of patients with idiopathic REM sleep behaviour disorder, there was autonomic dysfunction 5 years before the diagnosis of PD.29 Other autonomic features include erectile dysfunction, dizziness and hyperhidrosis, although these features usually do not predate the onset of PD.8 ,29 Disturbed thermoregulation and sweating are common prodromal features of PD or at presentation, although it is relatively unusual for disturbed sweating to lead to a medical consultation.
Neuropsychiatric features: Several case–control and prospective studies have shown a high prevalence of depression and anxiety in early PD and in the premotor phase of the disease. Depression occurs up to 10 years before PD diagnosis;28 ,30–32 studies suggest that 15% of patients with PD develop depression in the early stages of the disease or before diagnosis. Depression as a presenting feature of PD occurs in 2.5% of the cases and so this can be a PD chameleon.8 Another study showed increased incidence of PD in depressed adult patients.33 This is particularly common in elderly patients where the onset of severe depression in a patient previously unaffected by psychiatric disease can indicate emergent PD or dementia with Lewy bodies. Other non-motor symptoms include cognitive impairment, anxiety and lethargy.8
Motor symptoms
The asymmetry of PD means that some patients presenting with an apparent sudden onset illness may be erroneously diagnosed as having had a stroke.8 ,16 Atypical presentations can relate to both asymmetrical stiffness and sensory symptoms. Many patients describe their symptoms in terms of shoulder/arm pain and stiffness, and may develop secondary rheumatological problems, such as frozen shoulder34; this may lead to inappropriate or unnecessary treatment. Further unusual motor presentations include those related to asymmetrical motor performance in complex tasks and sports, including swimming and skiing. Exercise-induced dystonia occurs in about 20% of patients with early-onset PD and may be prominent in some patients with parkin disease.35
PD mimics
The most important PD mimics include tremor disorders, drug-induced parkinsonism, vascular parkinsonism and Parkinson's-plus conditions (box 3 and table 1). Patients with these diseases are often misdiagnosed as having PD. Evidence for the existence of PD mimics comes from functional imaging studies of patients who were initially diagnosed to have PD and from pathological follow-up of clinically diagnosed PD cases. These studies relate to both early-stage and late-stage diagnostic accuracy. The difficulties caused by PD mimics have recently been highlighted by the identification of patients recruited to PD treatment trials, passing apparently stringent diagnostic assessment for PD, who had normal functional dopaminergic imaging (ie, scans without evidence of dopaminergic deficits (SWEDDS)) and therefore, probably do not have PD.36–38 Movement disorders patients with SWEDDS represent a heterogeneous group of patients with a PD-like presentation. Possible explanations are dystonic and essential tremor, depression, vascular parkinsonism, psychogenic disease and dopa-responsive dystonia.38–40 The SWEDDS studies suggest that red flags for PD mimics include lack of true parkinsonian bradykinesia, occurrence of major dystonia and the clinical characteristics of the tremor.38 ,39 We will briefly outline the clinical features of the PD mimics.
Selected Parkinson's disease mimics
Tremor
-
Dystonic tremor
-
Isolated rest tremor
-
Indeterminate tremor
-
Essential tremor
-
Fragile X tremor–ataxia syndrome
-
Primary gait disorders
-
Progressive supranuclear palsy
-
Vascular parkinsonism/frontal gait disorder
-
Normal pressure hydrocephalus
-
Atypical parkinsonian disorders
-
Multiple system atrophy
-
Corticobasal degeneration
-
Dementia with Lewy bodies
-
Drug-induced parkinsonism
-
Neuroleptics
-
Dopamine blocking antiemetics
-
Sodium valproate
-
Depression
-
Early-onset and genetic parkinsonian disorders
-
Wilson's disease
-
Juvenile Huntington's disease
-
Spinocerebellar ataxias
-
Fronto-temporal dementia with parkinsonism
-
Pallido-pyramidal syndromes/neurodegeneration with brain iron accumulation
-
Clinical pointers and radiological features in parkinsonian syndromes
Tremor disorders
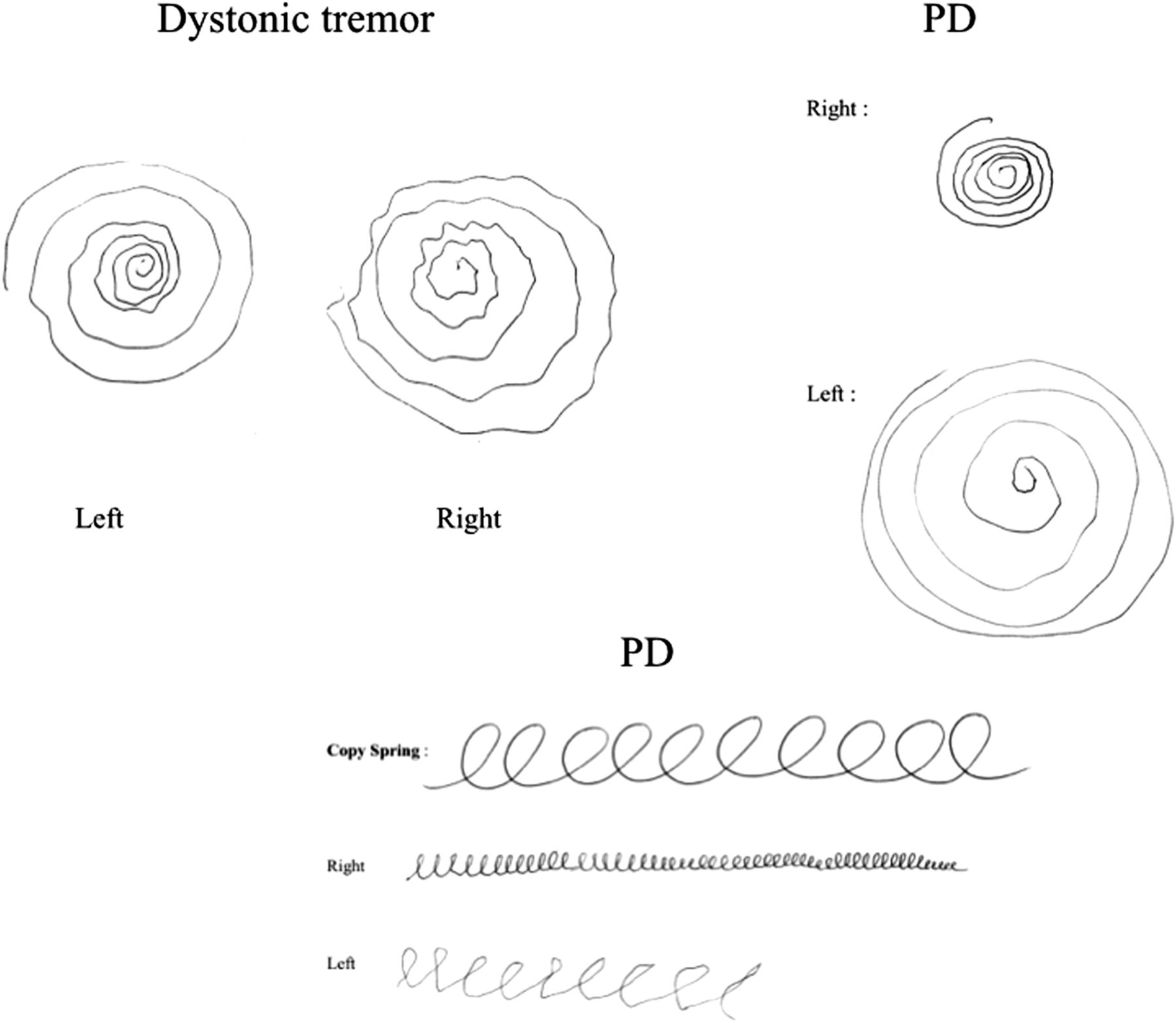
Patients with tremor commonly present to general neurology and movement disorder clinics: often the patient prompts the referral with concerns about possible PD. Most patients can be reassured after clinical assessment. Most patients with significant non-PD tremor who are referred to movement disorders clinics have dystonic tremor; however, there is some controversy on this point and diagnostic practices differ between the UK and other countries.41 Patients with both dystonic tremor and essential tremor frequently have action tremor and postural tremor rather than a rest tremor; some have a rest component, leading to diagnostic difficulties. Tremor assessment should include examination of patients in a relaxed position with arms supported, looking for rest tremor in sustained posture and during action. Rest tremor may only become apparent during other distraction tasks, such as counting backwards, walking or contralateral finger tapping.42 ,43 Action tremor may develop during finger-to-nose movement. We routinely examine patients’ handwriting, both in drawing Archimedes’ spirals and in writing a sentence44 (figure 1). Dystonic tremor may be particularly severe when pouring water from cup to cup. The clinical classification of tremor disorders—particularly those likely to be confused with PD—may be difficult, but clinical pointers are very important. Important features of non-PD tremor include a marked writing tremor and an action tremor more prominent than rest tremor.

Writing in Dystonic tremor and Parkinson's disease: Dystonic tremor – no micrographia and asymmetric tremor; Parkinson's disease – asymmetric progressive micrographia.
Dystonic tremor: The features of dystonic tremor include thumb extension tremor, jerkiness of tremor with flurries and task or position specificity. Patients do not have true parkinsonian bradykinesia and respond poorly to levodopa. Although concurrent dystonia may help, some patients with PD, particularly in early-onset disease, have prominent dystonia—with an extended thumb or ‘dinner-fork’ posturing—so this cannot be a definitive distinguishing feature. Dystonic tremor may be a major cause of disease in patients with SWEDDS. In patients with dystonic and essential tremor, dopamine-transporter scan (123I-FP-CIT SPECT scan (DaTscan)) is normal, indicating that there is no presynaptic dopaminergic deficit.39 ,40 ,45 ,46
Monosymptomatic rest tremor: Some patients present with a rest tremor that has the same clinical characteristics as PD tremor but without bradykinesia or without other features of PD. If this persists for more than two years, then this can be called ‘mono-symptomatic rest tremor’. Functional imaging in these patients may show dopaminergic denervation, suggesting that this condition is often a clinically restricted form of PD. These patients should not be diagnosed as having PD unless they develop bradykinesia, which in some patients never occurs or begins only after a long time delay.43 ,47
Indeterminate tremor disorder: According to the MDS Tremor Investigation Group classification, patients with indeterminate tremor satisfy the criteria for definite or probable essential tremor, but have either recognisable neurological disorders such as parkinsonism or have neurological signs of unclear significance, making the diagnosis of essential tremor doubtful.47
Essential tremor: This is probably the most common movement disorder, although many patients do not seek medical attention. Classical essential tremor is both action and postural, predominantly affecting the hands and is usually symmetrical. Essential tremor may be very mild. Classical essential tremor is relatively unlikely to be confused with PD, although essential tremor may be a potential cause of tremor in patients with SWEDDS.38 There may be a positive family history and beneficial response to alcohol.48–51
Fragile X tremor–ataxia syndrome (FXTAS) causes a progressive intention and postural tremor, with parkinsonism and cerebellar features. MR scan of brain shows cortical and subcortical atrophy with symmetrical lesions in the periventricular white matter and characteristically increased T2 signal in the middle cerebellar peduncle. FXTAS is caused by a pre-mutation expansion (55–200 repeats) of the CGG trinucleotide repeat in the fragile X mental retardation-1 (FMR1) gene. Although FXTAS can be mistaken for PD, it is more likely to be mistaken for multiple system atrophy-cerebellar subtype or cerebellar ataxia.52 ,53
Gait disorders
PD rarely presents primarily with a gait disorder and falls, though this can occur, particularly in the elderly. Presentation with a gait disorder suggests an alternative diagnosis. Close attention to posture, neck position and movement, arm position and arm swing is very helpful in diagnosing PD and its mimics. Patients with PD usually have a flexed trunk and neck posture with reduced arm swing and flexed elbows and a reduced step size. Normal or erratic step size, extended neck postures and normal or exaggerated arm swing are red flags, suggesting alternative diagnoses.
Progressive supranuclear palsy: This tauopathy is a primary disorder of gait and balance, as well as an atypical parkinsonian disorder. The gait disorder is characterised by a normal step size and erect, sometimes hyperextended posture, often with a lurching gait. In the early disease stages, patients with progressive supranuclear palsy have difficulty in standing and an uncontrolled descent when sitting. The typical form of the disease (progressive supranuclear palsy–Richardson's syndrome) is distinctive, involving a symmetric akinetic–rigid syndrome, axial rigidity, supranuclear gaze palsy, continuous frontalis overactivity, reduced blink rate, retrocollis and pseudobulbar palsy. The bradykinesia involves small-amplitude finger taps without decrement.54 In the early phases of the disease, there may be slowing rather than restriction of saccadic eye movements. Some forms of progressive supranuclear palsy—diagnosed pathologically—more closely resemble PD in the early disease stages (progressive supranuclear palsy–parkinsonism) and show an initial moderate response to levodopa.16 ,55 ,56
Vascular parkinsonism/frontal gait disorder: Vascular parkinsonism/diffuse cerebral small vessel disease is the most common cause of a frontal gait disorder. The diagnosis is based on the clinical features and the exclusion of normal pressure hydrocephalus, structural frontal lesions and conditions such as progressive supranuclear palsy. Frontal gait disorders (‘gait apraxia’) usually involve a short-stepped gait, without upper body parkinsonism, usually with an erect posture and normal or exaggerated arm swing. Patients may have asymmetrical difficulty in positioning the foot or leg when walking. Some patients have gait freezing when standing or turning. The management is focused mainly on treating vascular risk factors and providing supportive therapy for mobility and falls prevention. High-dose levodopa helps some patients with vascular parkinsonism, although this has not been formally evaluated in randomised controlled trials. As with PD, patients with vascular parkinsonism can respond well to auditory and visual cues and verbal instructions.57–59
Normal pressure hydrocephalus causes a similar clinical syndrome to vascular parkinsonism. Patients present with progressive cognitive decline, subcortical dementia, urinary incontinence and a frontal gait disorder. Brain imaging shows ventriculomegaly, disproportionate to the degree of cerebral atrophy. The diagnosis is confirmed by cerebrospinal fluid (CSF) removal or by dynamic CSF studies, such as a lumbar infusion test or a lumbar drain. CSF removal may improve gait, cognition or both. Some patients may benefit from a permanent CSF diversion procedure. Around 70% of these who undergo CSF diversion procedures show some initial gait improvement, but this often subsequently declines. Only one-third have sustained improvement at 3 years; the cognitive and urinary function generally has a worse long-term outcome than does gait.60–62
Atypical parkinsonian disorders
Multiple system atrophy: This synucleinopathy can be difficult to distinguish from PD, particularly in the early disease stages. Patients with multiple system atrophy often have asymmetrical parkinsonian bradykinesia with a good response to levodopa and may develop levodopa-induced dyskinesia. They may develop a flexed trunk and neck posture, identical to PD. Although autonomic features and gait disturbance are typical of multiple system atrophy, they can occur in PD. However, the relationship between these symptoms and disease onset is important. In multiple system atrophy, urinary incontinence, postural hypotension and gait disturbance are usually present at diagnosis or in the first few years after presentation, whereas in PD these occur later in the disease course (although constipation is often an early feature of both PD and multiple system atrophy). Useful additional clues to the diagnosis of multiple system atrophy include stretch-sensitive and action myoclonus affecting the fingers (mini-poly-myoclonus) and a predominantly cranio-cervical levodopa-induced dyskinesia often with dystonic facial movement and a ‘strangulated’ dysarthria.16 ,63 ,64
Corticobasal degeneration: This tauopathy presents with an asymmetrical akinetic–rigid syndrome (like PD). Loss of arm swing and unilateral stiffness often lead to an erroneous diagnosis of PD. There is often marked pain together with dystonia, apraxia, myoclonus and cortical sensory loss. Corticobasal degeneration does not respond well to levodopa treatment. At presentation, positional or postural limb instability, dyspraxia and myoclonus are important distinguishing features.65 ,66
Dementia with Lewy bodies: Dementia with Lewy bodies, PD and PD–dementia share common pathological and clinical features, and relate to a continuum of brainstem, limbic and neocortical Lewy body pathology. By definition, dementia with Lewy bodies involves major cognitive symptoms within the first year of onset, usually with parkinsonism. To some extent, the separation of these conditions is arbitrary and clearly some patients with dementia with Lewy bodies closely mimic those with PD. Prominent visual hallucinations and fluctuations in attention and awareness are important features in the diagnosis of dementia with Lewy bodies.67 ,68
Drug-induced parkinsonism
Various medications can cause parkinsonian symptoms and signs, including dopamine-depleting agents (such as tetrabenazine), dopamine antagonists (such as the antiemetics prochlorperazine and metoclopramide) and antipsychotics (fluphenazine, haloperidol and the depot preparations). Sodium valproate has recently been described as causing parkinsonism. High-dose cinnarizine and flunarizine can cause parkinsonism. Adulterated medicines bought on the internet containing kava-kava (Piper methysticum) and reserpine can cause severe parkinsonism.2 Illegal psychostimulant drugs containing ephedrine with manganese have caused severe drug-resistant parkinsonism in Eastern European countries, related to cerebral manganese toxicity.69 Sometimes these drugs may unmask underlying PD. The parkinsonian effects of these drugs can take up to a year to resolve after cessation of the medication. The DaTscan is normal in drug-induced parkinsonism, primarily due to blockade of D2 dopamine receptors.2 ,45 ,69–71
Depression/psychiatric disease
Slowing of movement and motor retardation, related to severe depression, can lead to the erroneous identification of bradykinesia and parkinsonism. Depression has been highlighted as one of the possible causes of SWEDDS and obsessional slowness can result from obsessive–compulsive disorder.39 ,40
Early-onset and genetic parkinsonism
Parkinsonism can occur in a wide variety of early-onset neurological and neurodegenerative diseases. We describe some selected conditions that can lead to diagnostic confusion below.
Monogenetic PD
PD can be a genetic/familial neurodegenerative disease. Genetic investigations may be worthwhile in patients who present before the age of 40 years or who have more than one affected relative. Familial PD can be autosomal dominant, for example, due to coding mutations (PARK1) or duplications/triplications (PARK4) in the alpha-synuclein gene or due to the common LRRK2 G2019S mutation.72 In general terms, patients with autosomal-dominant PD are indistinguishable from patients with sporadic PD,73 although some mutations, particularly the alpha-synuclein E46K mutation and triplication, can present with early-onset disease and marked early dementia.74–77 Mutations in parkin (PARK2), DJ1 (PARK7) and PINK1 (PARK6) occur in patients with early-onset disease.78 Patients with mutations in these genes have been reported to have a long disease course, with prominent dystonia, levodopa-induced dyskinesia and neuropsychiatric features without dementia, although some studies have suggested that these features relate to age at onset rather than the genetic aetiology.78–80
Wilson's disease: This is a rare autosomal-recessive disorder caused by autosomal recessive mutations in the ATP7B gene, leading to copper deposition in the liver, the basal ganglia and other organs. It would be unusual for Wilson's disease to mimic PD directly, as age at onset, concurrent dysarthria, dystonia, proximal tremor and psychiatric features usually suggest the alternative diagnosis. Nevertheless, clinicians need a high degree of suspicion in younger patients with movement disorders involving parkinsonism; this should include a careful search for Kayser–Fleischer rings with slit-lamp examination and measurement of serum caeruloplasmin. It is important to consider Wilson's disease in young people presenting with any movement disorder and/or dysarthria.45 ,81 ,82
Huntington's disease: Juvenile Huntington's disease classically presents with parkinsonism; important clues are a positive family history and a concurrent fronto-behavioural syndrome.83
Spinocerebellar atrophy (SCA) types 2 and 3: SCA-2 and 3 have been described as presenting with a presentation typical of Lewy body PD with asymmetric parkinsonism and a good response to levodopa, particularly in non-Caucasian patients.84
Fronto-temporal dementia with parkinsonism: The major forms of fronto-temporal dementia relate to underlying tau, TDP-43 or FUS gene pathology, and all may present with parkinsonism. Fronto-temporal dementia with parkinsonism can be genetic, with mutations in the MAPT, GRN or C9ORF72 genes. A prominent frontal behavioural syndrome and positive family history are important clues as to the underlying diagnosis. FTDP-17 due to mutations in MAPT can present with both PD and progressive supranuclear palsy-like syndromes.85–87
Neurodegeneration with brain iron accumulation: This represents a group of disorders with early-onset progressive parkinsonism–dystonia, associated with iron deposition in the basal ganglia and characterised by low signal on T2-MRI. Some of these conditions may initially respond well to levodopa and lead to initial diagnostic confusion with PD. However, the appearance of atypical features such as early bulbar involvement, spasticity, pyramidal signs, dementia and supranuclear gaze palsy together with characteristic brain imaging appearances point to the underlying diagnosis. Pantothenate kinase-associated neurodegeneration accounts for 50% of neurodegeneration with brain iron accumulation. This is an autosomal-recessive condition due to a mutation of the PANK2 gene. MR brain imaging characteristically shows the ‘eye of the tiger’ on T2-weighted imaging with low signal in the pallidum surrounding a high-signal focus. Neuroferritinopathy is an autosomal-dominant condition with high penetrance due to mutation of the ferritin light chain gene (FTL1). This is a neurodegenerative condition characterised by progressive stereotyped dystonic movements with orofacial dyskinesia and normal cognition. The MR scan of brain shows cystic degeneration of the basal ganglia and the ‘eye of the tiger’ sign may also occur in this condition. Other neurodegeneration with brain iron accumulation syndromes include acaeruloplasminaemia due to mutation of the CP gene, infantile neuroaxonal dystrophy, PLA2G6-associated neurodegeneration, FA2H, C19orf12, ATP13A2 and other conditions.88–92
Imaging biomarkers in parkinsonism
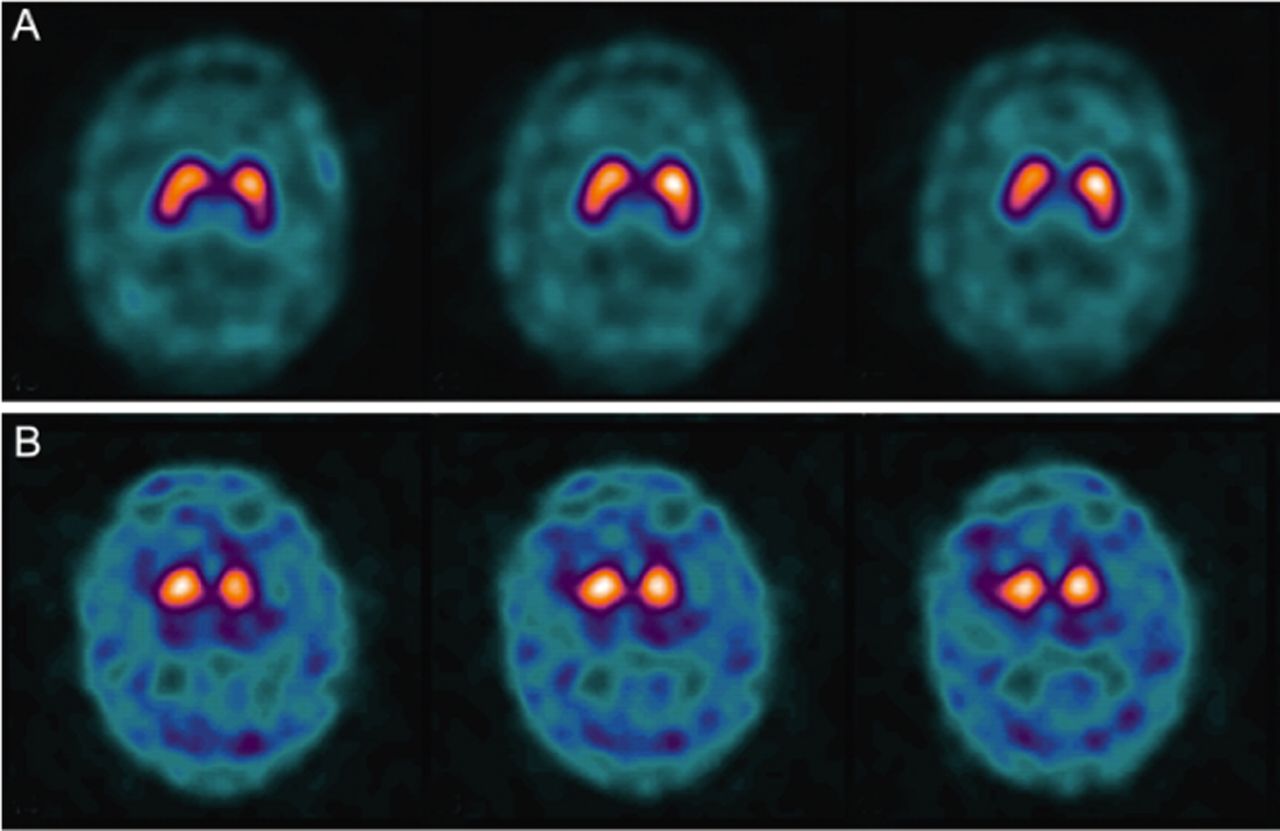
Although applying the QSBB criteria gives high sensitivity and specificity in detecting established patients with PD, applying this in the early stages of the disease is more difficult when the clinical picture might not fulfil the QSBB criteria (because of lack of supportive features related to progression, response to drug treatment or because true parkinsonian bradykinesia may be difficult to assess with prominent postural tremor). The routine use of neuroimaging in all patients with PD is not recommended.93 The National Institute of Health and Care Excellence (NICE) in the UK recommends the use of MR scan of brain in the differential diagnosis of parkinsonian disorders but not PD,93 and similarly, the American Academy of Neurology (AAN) guidelines suggest that MR scan of brain can help to distinguish PD from multiple system atrophy.94 In neurological practice, MR scan of brain may be useful for patients with atypical features for PD, for example, lower body-predominant parkinsonism, strictly unilateral disease, lack of asymmetry, early urinary disturbance and early cognitive impairment. MRI particularly helps in patients with complex parkinsonian disorders—older patients may have markers of progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration and vascular disease58 (table 1). MRI can detect specific abnormalities, such as pontine MRI T2 signal change (the ‘hot-cross bun’ sign) and reduced MRI T2 signal of the putamen in multiple system atrophy (figure 2). Midbrain, and superior cerebellar peduncle atrophy in progressive supranuclear palsy (figure 3).95 ,96 In younger patients, there may be structural basal ganglia abnormalities and/or brain iron deposition, in neurodegeneration with brain iron accumulation disorders. Functional imaging of the dopamine transporter using SPECT and (123I) ioflupane as a tracer (DaTscan) is well established in clinical practice as conditions such as PD, which lead to nigrostriatal dopaminergic denervation, or very rarely to loss of the dopamine transporter, lead to loss of the normal pattern on DaTscan97 ,98 (figure 4). The European Medicines Agency and Food and Drug Administration licences for DaTscan indicate that this tool can differentiate parkinsonian disorders from essential tremor, but that DaTscan does not differentiate PD from other parkinsonian disorders (such as progressive supranuclear palsy and multiple system atrophy). NICE and the AAN recommend a DaTscan to distinguish essential tremor from parkinsonian disorders.93 ,94 DaTscan is also normal in other conditions that may mimic PD, including drug-induced parkinsonism, psychogenic tremor, some central nervous system infections, hydrocephalus, pallidal atrophies, X-linked dystonia–parkinsonism and dopa-responsive dystonia. In specialist settings where clinicians suspect these rarer PD mimics, DaTscan may be useful.98 ,99 Using 123iodine-meta-iodobenzyl guanidine (123I-MIBG) myocardial scintigraphy, patients with PD have significantly lower heart-to-mediastinum average count ratio than atypical parkinsonian disorders, reflecting postganglionic sympathetic cardiac denervation in PD.100 NICE and AAN guidelines do not currently recommend using this technique.93 ,94

T1 MR scan of brain (axial) showing the ‘hot-cross bun’ appearance in a patient with multiple system atrophy.

T1 MR scan of brain (sagittal) showing the ‘hummingbird’ sign in a patient with progressive supranuclear palsy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
DaTscan: dopamine-active transporter (DaT) scans showing binding of 123I-fluoropropyl (FP–CIT) to DaT protein in the nigrostriatal nerve endings of the striatum. (A) Normal: symmetrical normal specific binding of FP–CIT in striatum. (B) Parkinson's disease: reduced specific binding of FP–CIT in the posterior striatum, particularly on the left (picture courtesy of P Kempster45).
Further imaging approaches are in development on a research basis. 7-T MR scanning can detect structural nigral abnormalities.101 Diffusion-tensor MRI and diffusion-weighted imaging have shown some promise in diagnosing PD and its mimics.99 Transcranial sonography can detect hyperechogenicity in the midbrain of patients with PD. Although this is not specific, it may be a marker of susceptibility to PD.99 ,102 ,103
Conclusions
Over the last 20 years, the diagnosis of PD has greatly improved through the use of clinicopathological criteria, improved knowledge and understanding of non-PD movement disorders and through our improved understanding of the non-motor features of PD. It is likely that, in the coming years, imaging or fluid biomarkers will assist the clinical diagnosis of PD, particularly in the early stages. However, at present, the most important aspects of the diagnosis of PD and the identification of PD chameleons and mimics remain the careful observation of the evolution of symptoms, signs and treatment response, and vigilance for new and atypical clinical features.
Acknowledgments
We acknowledge the contribution of Dr Simon Lewis and Professor Andrew Lees, whose comments greatly strengthened this article.
References
Footnotes
-
Contributors KA and HRM wrote the article. KA carried out the literature search and wrote the first draft. HRM revised the article and both are responsible for the final version.
-
Funding HRM is supported for work on PD by Parkinson's UK (grant 8047) and the Medical Research Council UK (G0700943, G1100643).
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Simon Lewis, Sydney, Australia, and Andrew Lees, London, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Improving the diagnostic accuracy in parkinsonism: a three-pronged approach
- Sleep disturbance in movement disorders: insights, treatments and challenges
- Is it Parkinson’s disease, and if not, what is it?
- Clinical utility of dopamine transporter single photon emission CT (DaT-SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes
- Parkinson’s disease: clinical features and diagnosis
- The role of DAT-SPECT in movement disorders
- Systematic clinical approach for diagnosing upper limb tremor
- Open questions on the nature of Parkinson’s disease: from triggers to spreading pathology
- How valid is the clinical diagnosis of Parkinson's disease in the community?
- NOTCH2NLC-related disorders: the widening spectrum and genotype–phenotype correlation