Article Text

Statistics from Altmetric.com
Introduction
Transverse myelitis requires careful investigation, as although many causes respond acutely to immunomodulation, the longer term management depends upon its precise cause. We describe a patient presenting acutely with a corticosteroid-responsive longitudinally extensive transverse myelitis and granulomatous lung lesions with a previous history of recurrent generalised lymphadenopathy, pyogenic infections and idiopathic thrombocytopenic purpura. We initially suspected an underlying primary autoimmune disorder. However, investigations showed granulomatous common variable immunodeficiency (gCVID) and following regular intravenous immunoglobulin, her symptoms did not recur. We discuss this relatively common immunodeficiency disease, frequently misdiagnosed as a systemic autoimmune disease (often sarcoidosis). We also discuss the range of neurological syndromes, including transverse myelitis, that may accompany common variable immunodeficiency (CVID).
Case report
A 41-year-old Caucasian woman presented with a 1-week history of progressive bilateral leg weakness, with urinary hesitancy and frequency. She had an intriguing past medical history of multiple episodes of corticosteroid-responsive widespread tender lymphadenopathy over 10 years, splenectomy for refractory idiopathic thrombocytopenic purpura in 2005, frequent respiratory tract infections and multiple buttock abscesses.
On examination at presentation, there was increased tone in both lower limbs with mild bilateral proximal weakness. Her reflexes were pathologically brisk in both legs and plantars were extensor. There were no sensory abnormalities. Upper limb and cranial nerve examinations were normal.
Routine blood tests were normal, including full blood count, renal function, liver function, clotting function and inflammatory markers. Her MR scan of brain was normal, but an MR scan of spine showed longitudinally extensive cord signal change between T2 and the conus medullaris (figure 1) with focal contrast enhancement at T6/7 (figure 2), consistent with transverse myelitis. Further investigation was aimed at finding the cause from a wide differential (table 1).
Causes of longtitudinally extensive transverse myelitis

T2-weighted sagittal MR scan of spine showing longitudinally extensive cord signal change between T2 and the conus medullaris.

Post-contrast T1-weighted sagittal MR scan of spine showing focal enhancement at T6/7.
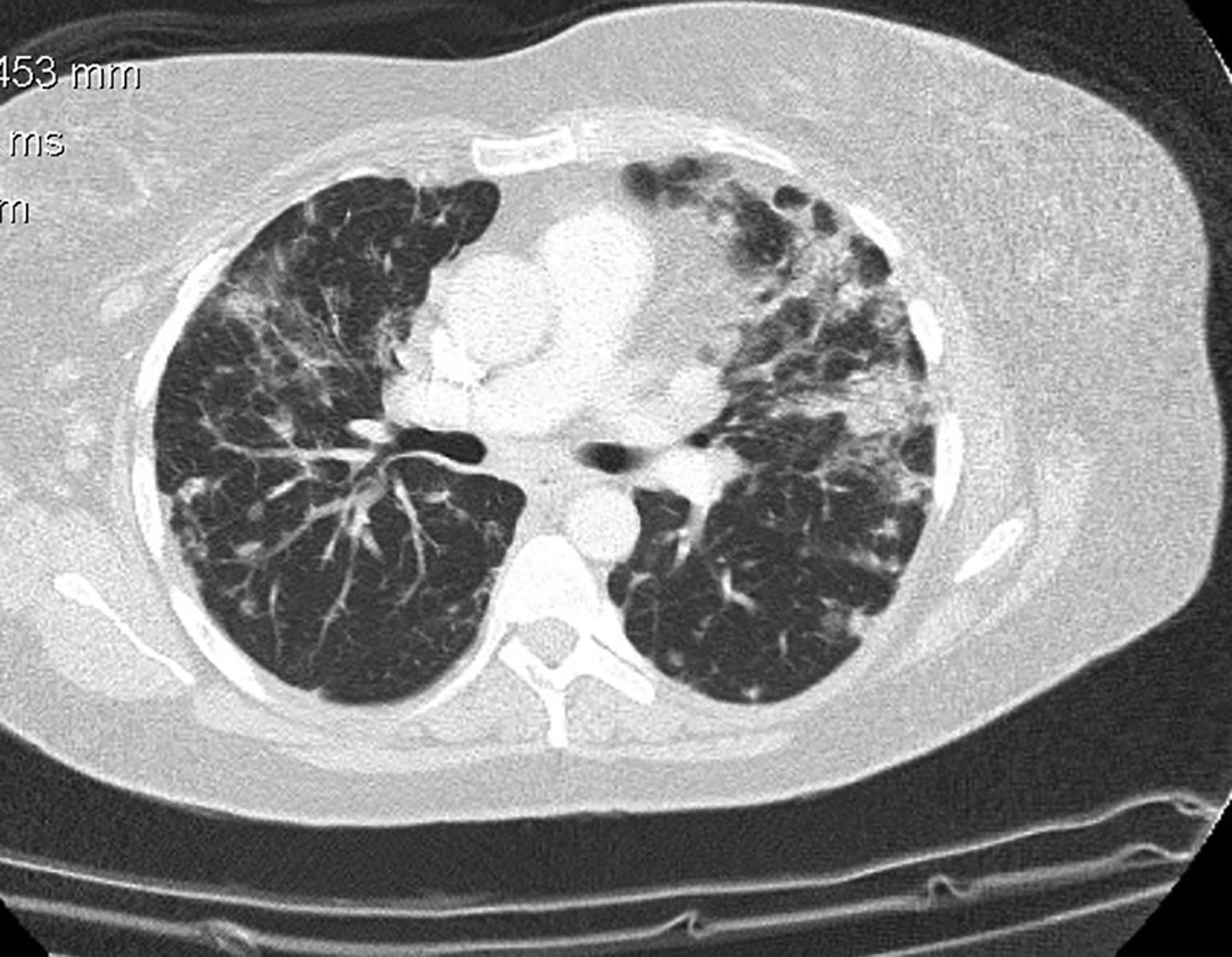
Her cerebrospinal fluid (CSF) was acellular with a mildly elevated protein of 0.56 g/L (0.15–0.45), normal glucose and negative oligoclonal bands. Contrast-enhanced CT imaging showed extensive mediastinal and abdominal lymphadenopathy with bronchiectactic and nodular changes throughout both lung fields (figure 3), raising the possibility of sarcoidosis, although her serum ACE was normal at 33 U/L (25–82). Subsequent lymph node biopsy showed no evidence of lymphoma or granulomatous disease but did find necrotising lymphadenitis with CD68 histiocytes. Biopsy immunostaining was negative for Brucella, Trichinella, hydatid and toxoplasma. A transbronchial biopsy with bronchial washings showed only non-specific chronic inflammation. Other negative investigations included: HIV serology and p24 antigen; Mantoux and QuantiFERON-TB test; autoimmune profile, including antinuclear antibodies, dsDNA, extractable nuclear antigen and anti-aquaporin-4 antibodies and blood/CSF testing for bacterial and viral organisms. including varicella zoster, Lyme and Brucella by PCR. Immunoglobulin testing showed a low level of IgG at 1.8 g/L (6.0–13.0), IgA at 0.34 g/L (0.8–3.0) and normal of IgM at 0.44 g/L (0.4–2.5).

{kind=link}
{kind=link}
{kind=link}
Post-contrast axial CT scan of chest showing bilateral lung nodules and traction bronchiectasis.
She responded well to a 3-day course of high-dose intravenous methylprednisolone and fully recovered from her neurological symptoms after rehabilitation. Repeat MR scan of cord some months later showed less intrinsic cord signal change and no focal enhancement.
Further investigation of her hypogammaglobulinaemia, including lymphocyte subsets (normal CD4 and CD8 T cells, minimally elevated CD19 B cell and CD56 NK cell numbers), B lymphocyte subpopulations and functional antibody responses to immunisations, suggested a diagnosis of CVID (granulomatous form). Peripheral blood B cells showed a phenotype typical of gCVID with low (0.2% of lymphocytes) switched memory B cells (CD19, CD27, IgM−), normal range >2%, with a high proportion (52.5%) of B cells expressing CD21 (normal range <10%), a marker of autoimmunity in CVID. There were inadequate antibody responses to test vaccinations with Prevenar 13 (Pneumococcus) and tetanus toxoid. Haemophilus antibodies were protective but did not increase post-vaccination. The CD68 histiocytes seen previously on lymph node biopsy were a marker of activated polyclonal macrophages, an expected finding in gCVID.
She subsequently responded successfully to replacement dose (0.5 g/kg/3 weeks) of intravenous immunoglobulin and currently has had no recurrence of her neurological or systemic symptoms.
Discussion
CVID is the most common symptomatic primary immunodeficiency (table 2), with a prevalence between 1:2500 and 1:50 000 in the general population.1
Causes of immunodeficiency
The cause is unknown, but hypotheses include the role of disease susceptibility genes, such as transmembrane activator and calcium-modulator and cyclophilin-ligand interactor2 and subsequent environmental stimuli, such as cytomegalovirus infection.3 Its common clinical features (table 3) include autoimmunity and granulomatous inflammation, which may be misdiagnosed as sarcoidosis, especially when lymphadenopathy is present.4
Common features of common variable immunodeficiency
Recurrent lymphadenopathy and bacterial infections coupled with hypogammaglobulinaemia and a coexisting autoimmune history (idiopathic thrombocytopenic purpura) raises the suspicion of an underlying diagnosis of CVID. The diagnosis is criteria based, involving low immunoglobulins and impaired functional antibody responses.5 Therefore, a diagnosis of granulomatous form of CVID is an ill-defined subset of this condition. This is further supported by the fact that all CVID patients can have granulomatous, autoimmune and inflammatory phenomena, which are all probably inter-related and the consequence of a dysfunctional immune response to persistent infection. Immunoglobulin replacement prevents infections and currently is the mainstay of therapy for CVID.5 Immunoglobulin replacement does not reliably reverse autoimmune or inflammatory complications and so corticosteroids or other immunosuppressive drugs are required, although evidence of benefit is lacking.5
Neurological features in CVID are rare. However, several reported cases, taken together, suggest that this is a true association. Initial cases of transverse myelitis associated with CVID occurred among a case series of encephalomyelitis in primary hypogammaglobulinaemia.6 Subsequent cases of transverse myelitis have also shown this association, as well as other neurological manifestations such as cognitive impairment, ataxia, spasticity, weakness7 and bilateral optic neuritis.8 As in this case, almost all of the patients reported previously responded well clinically to a combination of corticosteroids and intravenous immunoglobulin with no recurrences. Hypotheses for neurological involvement in CVID include a possible autoimmune reaction against neuronal tissue,7 a yet undefined infectious agent7 and Vitamin E deficiency due to an associated enteropathy,9 although none have yet been proven. This is evidently an area of ongoing research with further surveillance of CVID patients recommended. Furthermore, myelitis is a recognised feature of secondary immunodeficiency states such as HIV. However, its association with other primary immunodeficiency syndromes has been limited to infective causes, such as coxsackie virus,10 secondary to the immunodeficient state rather than a direct consequence of the condition itself.
With regards to treatment, intravenous immunoglobulin and corticosteroid-related failures are rare in CVID. This has led to increasing interest in the role of the pro-inflammatory cytokine, tumour necrosis factor (TNF)-α and the use of TNF-α inhibitors such as infliximab. In relation to neurological involvement in CVID, infliximab has had mixed results. Reports suggest that a synergistic effect of intravenous immunoglobulin and infliximab improves neuroimaging appearances as well as systemic features such as fever, night sweats and lymphadenopathy whilst having no effect clinically on neurological deficits.11 Furthermore, a recent case of recurrent myelitis in CVID showed a good response to once weekly subcutaneous immunoglobulin with remission at 3-year follow up.12
References
Footnotes
-
Contributors EJ was the lead writer of the case report. RS reviewed and revised each draft. He also follows the patient up in the outpatient Neurology clinic. CRM was the Neurology registrar who initially reviewed and regularly provided Neurology input, while the patient was an inpatient. He also reviewed and revised the final draft. HL investigated and diagnosed the patient with CVID as an outpatient. She also contributed to the writing of the CVID section of the discussion.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned. Externally peer reviewed. This paper was reviewed by Neil Scolding, Bristol, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Recurrent myelitis in common variable immunodeficiency successfully managed with high-dose subcutaneous immunoglobulin
- Coexistence of pan-hypogammaglobulinaemia and primary ciliary dyskinesia
- Management of a pregnant woman with common variable immunodeficiency and previous reactions to intravenous IgG administration
- Autoimmune haemolytic anaemia due to immunodeficiency
- Genes associated with common variable immunodeficiency: one diagnosis to rule them all?
- Uncommon presentations of common variable immunodeficiency
- Does rituximab aggravate pre-existing hypogammaglobulinaemia?
- Response of refractory immune thrombocytopenic purpura in a patient with common variable immunodeficiency to treatment with rituximab
- Inflammatory bowel disease-like colitis pathology in a patient with common variable immune deficiency
- T-cell abnormalities in common variable immunodeficiency: the hidden defect