Article Text

Statistics from Altmetric.com
Introduction
Huntington's disease (HD) is an inherited neurodegenerative disorder characterised by a combination of motor abnormalities (chorea, dystonia, hypokinesia), cognitive impairment and neuropsychiatric symptoms, including depression, irritability and apathy. The age at onset is typically 35–45 years but it can present in juveniles and the elderly.1 The disease slowly progresses over 15–20 years.1 It is inherited as an autosomal dominant trait, with each child of an affected parent having a 50% risk of carrying the gene mutation and therefore of developing HD themselves. The cause is a CAG trinucleotide expansion mutation in the HTT gene that encodes the ubiquitously expressed huntingtin protein. Genetic testing by direct mutation analysis has been available clinically since the mutation was identified in 1993.2 Measurement of the CAG repeat length acts as a trait marker rather than a state marker; that is, the genetic test can be used either to confirm a clinical diagnosis of HD in symptomatic people or to predict whether or not an at-risk person will go on to develop HD. An abnormal result indicates that the person will one day develop HD, but gives no indication of his or her clinical state at the time of testing.
The pretest counselling requirements differ for diagnostic and presymptomatic (predictive) testing. The variety and complexity of clinical situations where diagnostic genetic testing for HD may be indicated make it impractical to construct guidelines such as those governing predictive testing.3 ,4 The purpose of this article is to draw the attention of neurologists and psychiatrists to the challenges and pitfalls encountered in diagnostic testing for HD. We make the following recommendations to guide practice and help ensure the best possible outcome for individuals and their family.
A diagnostic genetic test is carried out where an individual is already symptomatic. According to current convention, the symptoms must include unequivocal motor signs. Neurologists (or neuropsychiatrists) are generally the professionals responsible for the diagnosis of HD. Mood disturbance, personality changes or cognitive impairment and a positive family history, without motor signs, are not sufficiently specific to justify a diagnostic genetic test. Testing in such circumstances where the patient wishes to know his or her genetic status is a predictive test, requiring referral to a genetic counselling unit.
A predictive genetic test is an option for people at risk of developing HD. It provides information about whether someone will develop symptoms in the future, and, according to internationally agreed guidelines, can only be requested by a genetic counselling unit.4 Pretest counselling is an essential requirement to allow a patient adequate opportunity to reflect on what will be an irrevocable decision.
Diagnostic genetic testing
Confirming the clinical diagnosis of an inherited disorder has consequences for the patient and other family members (figure 1). Careful attention to timing and needs of the family can maximise the potential therapeutic value of the diagnostic process. For example, a diagnosis sometimes helps to explain worrying symptoms and thus comes as a relief to the patient and his or her family; however, a diagnosis of HD can induce or worsen existing depression: the risk of suicide and suicidal ideation increases just before and after disease onset.5 ,6 Where a diagnosis is confirmed, the patient's partner, children, grandchildren and even siblings may need a lot of information, especially if there is no previously known family history of HD.
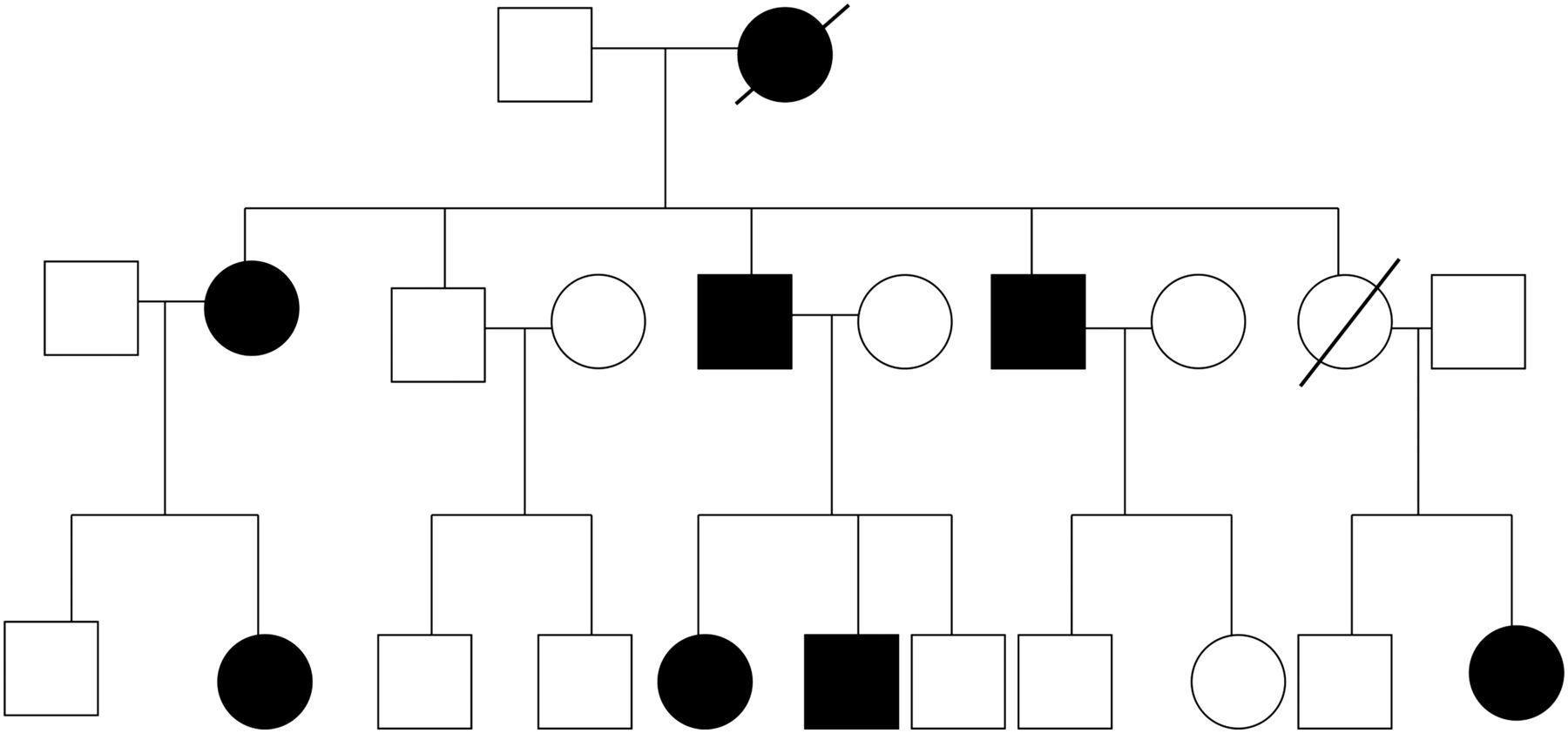
{kind=link}
A typical pedigree for a family affected with Huntington's disease (HD). The autosomal dominant mode of inheritance can lead to multiple cases over many generations, so an unfavourable test result often has serious implications for many other family members.
When should we consider diagnostic genetic testing
The patient with a positive family history and specific motor symptoms
In this situation, the clinical diagnosis is not difficult; the patient is usually aware of symptoms, and the test becomes little more than a formality. However, even this can be more complicated than it first appears. Many patients with early HD are unaware of the involuntary movements and the early behavioural changes occurring around the time of onset.7 ,8 Some people have a striking lack of insight into their symptoms, which can persist until the disease is quite advanced.9–12 Commonly, the patient's spouse or another family member has suspected the diagnosis for some time before the patient became aware of symptoms. In some cases, a family member may even have prompted the referral, while the patient remains convinced that he or she is perfectly well. Such a person is unprepared for the diagnosis and may not be able, in the fullest sense, to give informed consent. The entire process of imparting the diagnosis and performing a confirmatory test requires care and sensitivity to avoid causing unnecessary distress and to avoid driving the person into a state of entrenched denial, which may hinder subsequent attempts at symptomatic treatment. It is, therefore, important to explore the patient's understanding of the situation before proceeding to genetic testing; extra time spent on this can save a great deal of trouble later. An additional appointment and/or offering additional support before diagnostic testing may help.
The patient with no family history, but specific symptoms likely to be HD
Diagnostic genetic testing is most clinically useful where a patient has typical symptoms and signs (including motor abnormalities) of HD but has no known family history. However, it is also the situation where the diagnosis—once the implications have been fully absorbed—may cause the most distress for both immediate and extended family. HD will not be the only differential diagnosis. Where HD seems possible, the neurologist may suggest proceeding with genetic testing. It is best to inform both the patient and immediate relatives about the hereditary nature of the disease before carrying out the test. If the diagnosis is confirmed, the clinician must recognise the needs of the entire family as well as the index patient and ensure that family members can access appropriate genetic counselling services.
The patient with a positive family history and prodromal symptoms, which suggest the impending onset of HD
The current diagnostic criteria for HD still require motor abnormalities such as choreiform movements. Although chorea can sometimes be the presenting symptom, many patients with HD experience prodromal behavioural changes such as irritability, anxiety or depression before motor onset,13 ,14 while others present with cognitive changes, often manifesting as difficulties in the workplace. Sometimes, these prodromal changes continue for a decade or more before the early motor signs appear. An unfavourable genetic test result in such circumstances does not exclude the possibility of a concurrent psychiatric disorder that is essentially unrelated to HD and thus cannot confirm the diagnosis. In such circumstances, it is preferable to adopt a tailored approach according to the patient's symptoms and key concerns. If the patient is not asking about his or her genetic status, it is appropriate to treat the presenting symptoms, for example, with selective serotonin-reuptake inhibitors for an existing depressive episode and arrange follow-up. Alternatively, if the patient finds it difficult to manage the uncertainty of not knowing his or her genetic status, as well as dealing with any symptoms, it would be appropriate to refer to the nearest genetic unit.
A major affective disorder or a psychotic condition such as schizophrenia appears more common in people who carry the HD gene mutation than in the general population; both conditions occur more frequently in some HD families than in others.15 ,16 This suggests that the HD mutation may exacerbate the risk of depressive disorder or schizophrenia in those people carrying an increased genetic susceptibility to depression or psychosis, by virtue of a distinct family history of one of these conditions. The increased risk of depression or psychosis occurs not only in the immediate prodrome of HD, but may occur decades before the onset of motor symptoms. Again, an unfavourable genetic test result in this instance does not exclude a concurrent psychiatric disorder essentially unrelated to HD and thus cannot be regarded as confirming the diagnosis. Diagnostic genetic testing for HD is inappropriate in such circumstances and patients requesting this should be referred for predictive testing. Genetic units usually work in close collaboration with colleagues in psychiatry in such circumstances.
While the principles outlined above usually work well, there are occasional situations where a patient investigated for cognitive impairment presents with a neuropsychological test profile entirely consistent with HD, but has only non-specific motor changes insufficient to justify a confident clinical diagnosis of HD. In such a case, where HD seems the most likely diagnosis, genetic testing is entirely reasonable.
The child with a family history of HD and features of juvenile HD
Juvenile HD (onset ≤20 years) poses particular diagnostic challenges. The phenotype may differ from the typical adult onset in that there may be more prominent poverty of movement and speech disturbance, especially in those rare cases with onset ≤10 years. Criteria proposed by Nance for diagnostic testing in those with onset ≤10 years are:17
-
a known family history of HD (often, but not exclusively, the father)
and two or more of
-
declining school performance
-
seizures
-
oral motor dysfunction
-
rigidity
-
gait disturbance.
Most juvenile HD cases, however, present in the second decade, with a similar clinical picture, but the typical ‘adult onset’ presentation is also possible. As with adult cases, there is no issue in arranging a genetic test if the child or young person has unequivocal neurological signs and a known family history of HD. There can be problems if the family history is unknown and, in exceptional cases, if a child has signs before the parent. However, the insidious onset of HD means that the parents are usually aware of non-specific changes in their child18 ,19 and may have concerns about school performance or behaviour before motor onset, which may or may not turn out to be HD related. It is crucial to support the parents and provide an individualised plan for follow-up, for example, arranging a baseline neuropsychological assessment20 ,21 and possibly an MRI scan.21 Neither is diagnostic in itself, but may be repeated at a future time; the planned follow-up may reassure the parents that their concerns are being taken seriously and the clinician can judge whether the symptoms are progressing.
Checklist before diagnostic genetic testing
-
Has the test been requested by a neurologist (or neuropsychiatrist) with relevant experience?
-
Has the patient been appropriately counselled about the implications of genetic testing and possible test outcomes?
-
Has the neurological examination shown evidence of motor symptoms?
-
Have the patient's family or caregiver been included in discussions about the diagnostic genetic test and hereditary implications of HD?
-
Has the family been informed about the option of referral for genetic counselling?
-
Has appropriate informed consent for diagnostic genetic testing been obtained by the patient/legal representative/person with parental responsibility?
-
Is the laboratory to which the sample is being sent accredited and do they have experience of performing HD genetic testing?
-
Have full clinical details, contact details of the requesting clinician and evidence that consent has been obtained noted on the laboratory request card?
-
Has the timescale for expecting a result been made clear to the patient family and the laboratory?
-
Has a face-to-face appointment with the person requesting the genetic test been arranged to give the result?
Checklist following a genetic diagnosis of HD
-
Has a plan for follow-up been discussed with the patient and his/her family?
-
Is there opportunity to refer the patient to a specialist HD management clinic?
-
Have details of the lay organisation been provided?
-
Have options to participate in research been discussed?
-
Do the patient's relatives wish to be referred for genetic counselling?
Clinicians should always offer follow-up to the patient and his or her family and should consider referring to a specialist HD management clinic. The opportunity to participate in research such as the REGISTRY or Enroll-HD studies22 provide the patient with a sense of purpose and may also offer future opportunities to participate in clinical trials. Family members may want to access genetic counselling, and the neurologist should be aware of the nearest genetic counselling unit and be able to provide details of the lay organisation for support. We suggest that a multi-disciplinary approach to HD care—taking account of the needs and expectations of both the affected individual and their family—will meet with the greatest success.
ONLINE RESOURCES
-
International Huntington Association http://www.huntington-assoc.com
-
European Huntington's Disease Network http://www.euro-hd.net
-
HD Buzz; http://www.hdbuzz.net
-
Huntington Disease Youth Organisation http://www.hdyo.org
Acknowledgments
The authors are grateful to the European Huntington's Disease Network (EHDN), which is supported by the CHDI Foundation, a not-for-profit organisation dedicated to finding treatments for Huntington's disease. DC and RM also wish to acknowledge the support of the National Institute for Health Research (NIHR) Biomedical Research Centre in Manchester.
References
Footnotes
-
Contributors The original draft was written by Dr DC, Dr RM and Dr OQ following an outline suggested by Dr MF at a meeting of the Working Group. All the other authors were involved in critical review and editing of the manuscript.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Anne Rosser, Cardiff, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Huntington’s disease: diagnosis and management
- Running a neurogenetic clinic
- Huntington’s disease
- Incidence and mutation rates of Huntington’s disease in Spain: experience of 9 years of direct genetic testing
- Genetic testing in motor neuron disease and frontotemporal dementia: a 5-year multicentre evaluation
- Genetic testing and reproductive choice in neurological disorders
- Ten years of presymptomatic testing for Huntington's disease: the experience of the UK Huntington's Disease Prediction Consortium
- Cardiovascular genetics: the role of genetic testing in diagnosis and management of patients with hypertrophic cardiomyopathy
- New problems in testing for Huntington's disease: the issue of intermediate and reduced penetrance alleles
- The differential diagnosis of chorea