Article Text

Abstract
Guillain–Barré syndrome (GBS) and its variant, Miller Fisher syndrome (MFS) have several subtypes, together forming a continuous spectrum of discrete and overlapping syndromes. Such is the heterogeneity within this spectrum that many physicians may be surprised to learn that these disorders are related pathophysiologically, and therefore share certain clinical features. These include history of antecedent infection, monophasic disease course and symmetrical cranial or limb weakness. The presence of cerebrospinal fluid albuminocytological dissociation (raised protein, normal cell count), antiganglioside antibodies and neurophysiological evidence of axonal or demyelinating neuropathy also support a diagnosis in many cases, but should not be relied upon. Mimics of GBS and MFS can broadly be divided into those presenting with symmetrical limb weakness and those presenting with brainstem signs. MFS and the pharyngeal-cervical-brachial variant of GBS are frequently mistaken for brainstem stroke, botulism or myasthenia gravis, whereas Bickerstaff's brainstem encephalitis is often diagnosed as Wernicke's encephalopathy. Chameleons or atypical presentations of GBS-related disorders include: paraparetic GBS, bifacial weakness with paraesthesias, acute ataxic neuropathy, acute ophthalmoparesis, acute ptosis and acute mydriasis. Many neurologists may also not be aware that deep tendon reflexes remain present or may even appear brisk in up to 10% of patients with GBS. Correct diagnosis of GBS-related disorders helps to avoid unnecessary investigations and allows early immunotherapy if appropriate.
- NEUROPATHY
Statistics from Altmetric.com
Introduction
Guillain–Barré syndrome (GBS) is an umbrella term describing a heterogeneous group of related disorders, including Miller Fisher syndrome (MFS) and GBS subtypes (box 1).1 The common pathogenesis of these disorders is mirrored by several shared clinical features, including: history of antecedent infection, monophasic disease course and symmetrical cranial or limb weakness. Many neurologists believe that GBS only affects the peripheral nerves, but this is not always the case, as 10% of patients display normal or even brisk deep tendon reflexes during the disease course.2 ,3 The clinical presentation of GBS-related disorders is heterogeneous and the diagnosis may not be obvious at first. Here, we review the most important differential diagnoses (Mimics) for patients presenting with acute flaccid paralysis and brainstem syndromes and highlight some of the more unusual presentations (Chameleons) of GBS-related disorders.
Guillain–Barré and Miller Fisher syndromes and their subtypes
Guillain–Barré syndrome
-
Paraparetic variant*
-
Pharyngeal–cervical–brachial weakness*
-
Acute pharyngeal weakness*†
-
Bifacial weakness with paraesthesias*
-
Miller Fisher syndrome
-
Acute ataxic neuropathy†
-
Acute ophthalmoparesis†
-
Acute ptosis†
-
Acute mydriasis†
-
Bickerstaff's brainstem encephalitis‡
-
Acute ataxic hypersomnolence‡†
-
*Localised forms.
-
†Incomplete forms.
-
‡Central nervous system form.
The GBS spectrum
GBS has one principal variant, known as MFS. Five per cent of patients with MFS develop weakness during disease course, indicating that MFS and GBS form a continuum.4 GBS and MFS have been subclassified into several subtypes, which together form a continuous spectrum of discrete and overlapping syndromes, affecting the cranial nerves and the limbs (figure 1). Although neurophysiologically, GBS-related disorders can be divided into axonal and demyelinating types; a classification based on clinical criteria is more useful.1
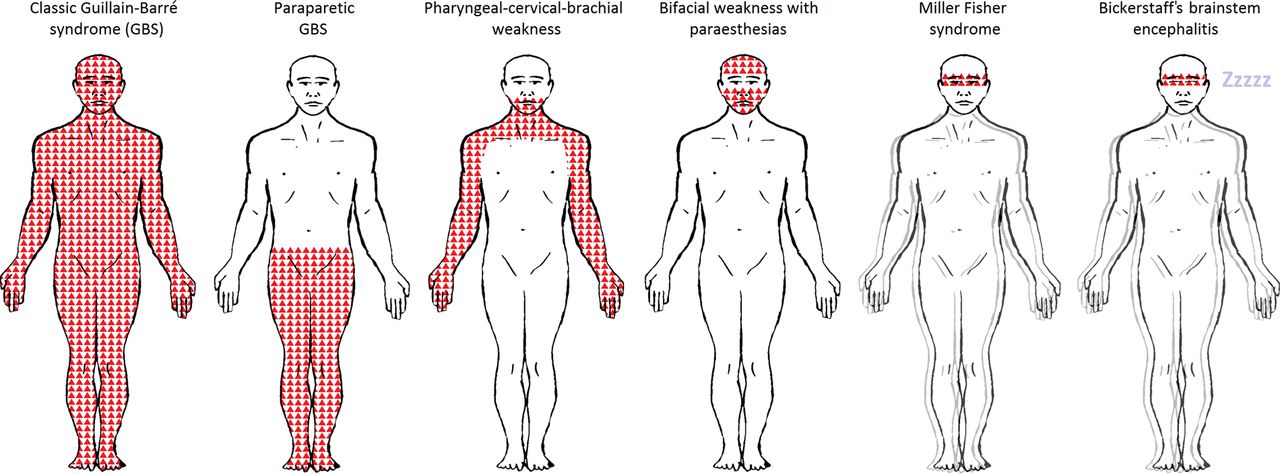
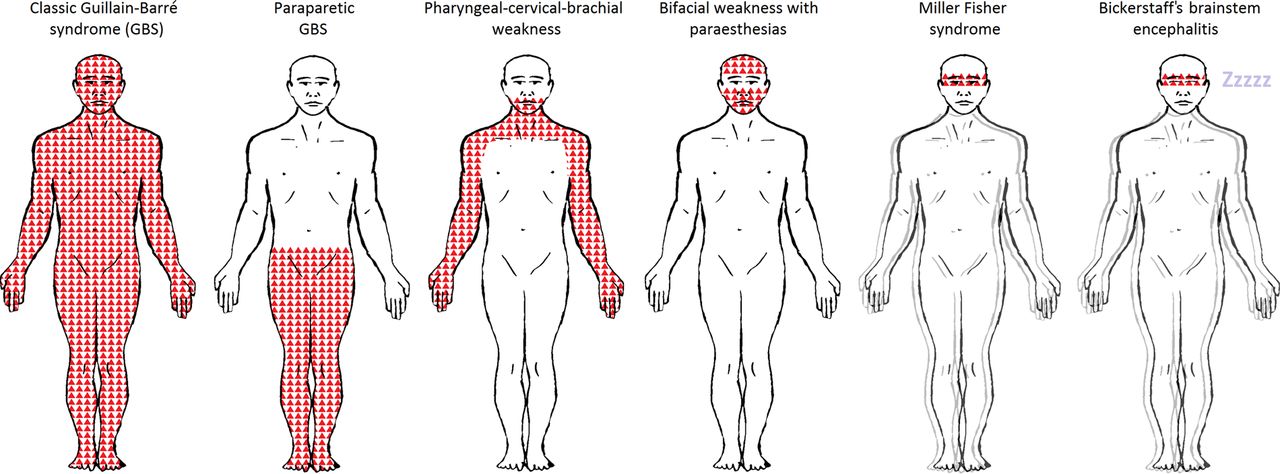{kind=link}
Patterns of weakness in Guillain-Barré syndrome (GBS) and Miller Fisher syndrome and their subtypes. GBS and Miller Fisher syndrome and their subtypes form a continuum of discrete and overlapping syndromes. Shaded areas indicate patterns of weakness. The double outline (blurring the figures) indicates the presence of ataxia. ‘Zzzzz’ indicates hypersomnolence. The pattern of weakness for each subtype are as follows: Classic GBS: tetraparesis with or without motor cranial nerve involvement; Paraparetic GBS: lower limbs; Pharyngeal–cervical–brachial weakness: bulbar, neck and upper limbs; Bifacial weakness with paraesthesias: facial; Miller Fisher syndrome: external ophthalmoplegia; Bickerstaff's brainstem encephalitis: external ophthalmoplegia. Facial weakness and motor cranial nerve involvement are more frequent in demyelinating-type classic GBS (acute inflammatory demyelinating polyradiculoneuropathy) than in axonal-type (acute motor axonal neuropathy). In Miller Fisher syndrome, there is ataxia and in its central nervous system subtype, Bickerstaff's brainstem encephalitis, there is additional hypersomnolence.
Patients with ‘classic’ GBS develop tetraparesis, although there are three localised subtypes. These include: the pharyngeal-cervical-brachial variant of GBS,5 causing bulbar, cervical and upper limb weakness; paraparetic GBS,6–8 causing bilateral lower limb weakness; and bifacial weakness with paraesthesias,9 causing bilateral facial weakness and distal limb sensory disturbance.
MFS is characterised by ophthalmoplegia, cerebellar-like ataxia and areflexia10 in the absence of limb weakness. Patients with additional involvement of the reticular formation, resulting in disturbance of consciousness, are said to have the central nervous system (CNS) subtype, known as Bickerstaff's brainstem encephalitis.11 There are also several incomplete forms of MFS, defined upon the presence or absence of ophthalmoplegia or ataxia. There is also overlap between the variants. For example, patients with ophthalmoplegia, ataxia and limb weakness are said to have MFS overlapped with GBS.
Clinical features
History
A careful history indicates that there are antecedent upper respiratory or gastrointestinal infective symptoms in over 90% of patients who develop GBS.12 Campylobacter jejuni, the most common cause of acute bacterial gastroenteritis, is consistently identified as the most frequent antecedent infection, occurring in up to 30% of patients.13 However, only one in 1000 patients with C. jejuni infection develop GBS.14 The median time interval between onset of diarrhoea and development of neurological symptoms is 10 days, but may be as short as 3 days or as long as 6 weeks.15 Several other bacterial and viral infections are associated with the development of GBS.16 These include: Haemophilus influenzae, Mycoplasma pneumoniae, cytomegalovirus, Epstein–Barr virus and varicella zoster virus. Some of the remaining patients with no antecedent infectious symptoms may still be harbouring asymptomatic infection. For example, half the patients with C. jejuni do not develop gastrointestinal symptoms.
There are also non-infective associations reported.16 These include parenteral gangliosides for treating peripheral neuropathy and various vaccines (eg, H1N1 influenza vaccine), which have the potential to induce molecular mimicry. Other associations include autoimmune diseases (eg, systemic lupus erythematosus), immunosuppressive drugs (eg, anti-TNFα therapy) and surgery, probably reflecting the increased susceptibility to known infective triggers, some of which are asymptomatic.
In most GBS and MFS subtypes, the onset of neurological symptoms is often with distal paraesthesia rather than weakness. This pattern also supports the presence of polyneuropathy. In our experience, many patients with GBS also report back pain, probably relating to inflammation of nerve roots.
Examination
There is a new diagnostic framework for GBS, MFS and their subtypes, which is likely to aid early diagnosis and classification in the future.1 The key examination finding in patients with GBS-related disorders is relatively symmetric weakness, although some patients do develop asymmetric and rarely unilateral weakness. What follows next is a brief overview.
Classic GBS (tetraparesis) can be diagnosed in patients presenting with bilateral flaccid limb weakness, and is highly likely when there is also facial weakness.17 Distal limb numbness, paraesthesias or pain, may be the initial presenting symptom. Weakness is often ascending and may involve respiratory muscles and cranial nerves. Up to 10% of patients have normal or exaggerated reflexes.2 ,3
MFS can be diagnosed in patients with ophthalmoplegia, ataxia and areflexia.10 Patients with isolated MFS have no limb weakness. Patients with additional hypersomnolence have Bickerstaff's brainstem encephalitis.11 There are also incomplete forms of MFS in which ophthalmoplegia or ataxia is absent.18–21
There may also be overlap between subtypes.1 Patients with MFS or Bickerstaff's brainstem encephalitis who develop limb weakness can be diagnosed as having overlap with GBS. Patients with prominent ophthalmoplegia or ataxia must have overlap with MFS. For example, patients presenting with pharyngeal-cervical-brachial weakness and ataxia have overlap with the pharyngeal-cervical-brachial variant of GBS, MFS or one of its subtypes.
Time course and progression
A core clinical feature of all GBS subtypes is the characteristic disease time course. In 90% of patients, this is monophasic, although up to 10% of patients develop recurrent or relapsing GBS.22 The time interval between onset of neurological symptoms and nadir ranges between 12 h and 28 days and is usually followed by subsequent clinical plateau or improvement.23 Treatment-related clinical fluctuations may occur within 8 weeks after starting immunotherapy and are regarded as part of a monophasic course. Patients who deteriorate after 8 weeks from initial onset or deteriorate three times or more despite appropriate immunotherapy should be diagnosed with acute-onset chronic inflammatory demyelinating polyradiculoneuropathy rather than GBS.24 Despite appropriate immunotherapy, the overall mortality in patients with GBS is 9% and 17% are left with severe disability.25 The prognosis in MFS is usually good.
Investigations
The most important early investigation is neuroimaging to exclude an inflammatory, ischaemic or structural cause for weakness, if felt clinically indicated. In the case of GBS, MR scans of brain and spinal cord should be normal, although there may be gadolinium enhancement of proximal nerve roots. In MFS and the pharyngeal-cervical-brachial variant of GBS, MR scan of the brain helps to exclude brainstem pathology, including restricted diffusion caused by acute stroke. Gadolinium contrast helps to exclude meningeal pathology, especially at the skull base. Abnormal signal change on T2-weighted MRI occurs in 11% of patients with Bickerstaff's brainstem encephalitis and may involve the upper mesencephalon, thalamus, cerebellum or brainstem.26 Typically, cerebrospinal fluid (CSF) shows albuminocytological dissociation (high protein, normal cell count). In one large series, CSF protein was raised in 49% of patients on day 1 and 88% after 2 weeks. Fifteen per cent had a mild pleocytosis (5–50 cells/µL (≤5)) but no one had more than 50 cells/µL.3 Although nerve conduction studies should be performed early in suspected GBS, they are often non-diagnostic in the first week and should not be relied upon, or delay treatment, if there is a high index of suspicion for GBS. Repeat CSF and nerve conduction studies after the first week are therefore invaluable if there is any doubt over the diagnosis. Serum antiganglioside antibody testing can help to support the diagnosis,27 but many centres test only for anti-GM1 and anti-GQ1b antibodies, and the results are often delayed.
Neurological conditions which mimic GBS, MFS and their subtypes
GBS, MFS and their subtypes have numerous mimics, which can make diagnosis difficult, especially in early disease. In this review, we highlight the most important differential diagnoses based on anatomical distribution of weakness, although there is significant overlap. For example, botulism may mimic MFS or pharyngeal-cervical-brachial weakness in early disease but GBS in more established disease.
Acute flaccid paralysis mimicking GBS
Although GBS is the most common cause of acute flaccid paralysis, the differential diagnosis is broad (box 2) and may involve any part of the motor system below and including the spinal cord. A detailed history provides clues to aetiology and should include: recent travel, antecedent infective symptoms (eg, upper respiratory tract), vaccinations, insect or animal bites (eg, tick or snake bites), exposure to toxins (eg, organophosphates), drugs or contaminated food (eg, pickled sausages in the case of botulism) or water, systemic symptoms (eg, fever, rash), trauma (eg, neck injury), family history (eg, familial hypokalaemic periodic paralysis) and psychiatric symptoms. By definition, patients presenting with acute flaccid paralysis do not show signs associated with CNS weakness (ie, increased muscle tone, hyper-reflexia, clonus and extensor plantar responses), but it is important to look for other features of central involvement including: cerebellar ataxia, change in behaviour or cognition and sphincter disturbance. Meningism, myalgia and autonomic instability also narrow the differential.
Box 2 Differential diagnoses for classic Guillain–Barré syndrome
Acute flaccid paralysis
Viruses targeting anterior horn cells or motor neurones
-
Poliomyelitis, non-polio enterovirus (enterovirus 71), West Nile virus
-
Herpes simplex virus, cytomegalovirus, Epstein–Barr virus, varicella zoster virus
-
Rabies virus, HIV
Transverse myelitis
-
Mycoplasma pneumoniae
-
Herpes simplex virus, cytomegalovirus, Epstein–Barr virus, varicella zoster virus
Spinal cord injury
-
Acute spinal stenosis (eg, disc prolapse, epidural abscess or haematoma)
-
Anterior spinal artery occlusion
Acute peripheral neuropathies
-
Infections (eg, herpes simplex virus, HIV)
-
Consumption of toxins or poisons (eg, puffer fish poisoning (tetrodotoxin), lead, thallium, arsenic)
-
Tick paralysis, Lyme disease
-
Porphyria
Neuromuscular junction disorders
-
Myasthenia gravis
-
Lambert–Eaton myasthenic syndrome
-
Botulism
Neuromuscular weakness related to critical illness
-
Critical illness neuropathy and myopathy
Muscle disorders
-
Acute myositis
-
Periodic paralysis
-
Functional
Here, we consider viruses targeting anterior horn cells and motor neurones, acute transverse myelitis, spinal cord injuries, acute peripheral neuropathies, neuromuscular weakness due to critical illness, disorders of the neuromuscular junction and disorders of muscle.28
Viruses targeting anterior horn cells or motor neurons
Certain viruses targeting anterior horn cells and motor neurones may cause acute flaccid paraparesis.29 Extensive necrotising myelopathy on MRI is associated with herpes simplex virus and West Nile virus. Poliomyelitis should be considered in unvaccinated individuals, especially those with recent travel to endemic regions.30 Most patients infected with polio remain asymptomatic, but 0.1%–1.0% develops paralysis. Typically, there is a prodromal flu-like illness, with fever, neck stiffness and significant myalgia. Rapidly progressive, often asymmetric paralysis, affecting proximal more than distal muscles, develops within 48 h in the absence of sensory deficit. CSF examination is usually lymphocytic.
Non-polio enterovirus, especially enterovirus 71, may cause acute flaccid paraparesis and there have been recent outbreaks in Australia and Cambodia.31 Typically, a 1–2 week prodromal illness of diarrhoea, lethargy, irritability and nuchal rigidity is followed by acute flaccid paraparesis and other neurological symptoms in up to 20% of individuals. The mortality is high.
Rabies virus may cause acute flaccid paraparesis 1–2 months after initial exposure.32 Early symptoms include behavioural changes and autonomic instability, followed by ascending paralysis associated with sphincter involvement and sensory disturbance.
Direct involvement of the spinal cord, nerve roots and peripheral nerves may occur in HIV or AIDS, but more often acute flaccid paraparesis is caused by the effects of opportunistic infections. These include cytomegalovirus, Epstein–Barr virus, varicella zoster virus and herpes simplex virus, which may also trigger acute motor axonal neuropathy.16 Cytomegalovirus and, less commonly, herpes simplex virus 2 or varicella zoster virus also cause polyradiculoneuropathy, especially if the patient is immunocompromised. Typically, there is ascending leg weakness, perineal paraesthesia, leg pain and variable bladder involvement. CSF usually shows increased polymorphs and protein with reduced glucose. Neurophysiologically, there is evidence of axonal neuropathy. Clinicians should also consider other infections, including syphilis, toxoplasmosis, giardiasis and tuberculosis.29 Patients with AIDS with cachexia may develop rapidly progressive weakness due to profound B12 deficiency.
Acute transverse myelitis
Acute transverse myelitis may cause acute flaccid paraparesis, usually with radiological evidence of spinal cord involvement. Initial spinal shock is followed by acute flaccid paraparesis associated with bladder disturbance and a sensory level. Often patients present with back pain. There are several infectious agents implicated, including—but not limited to—M. pneumoniae, herpes viruses, rabies virus, hepatitis A virus, enteric fever and parasitic infections (eg, schistosoma).28 ,29
Spinal cord injury
Spinal cord injury can usually be elicited from the history, and one should always ask about recent falls. Epidural abscess or haematoma may cause acute spinal cord compression and is usually associated with focal pain and can be shown radiologically. Anterior spinal artery occlusion may cause abrupt onset of acute flaccid paraparesis and should always be considered postoperatively if there has been cardiothoracic surgery requiring aortic clamping.
Acute peripheral neuropathies
In addition to GBS, clinicians should consider other causes of acute peripheral neuropathy. Acute-onset chronic inflammatory demyelinating polyradiculoneuropathy is difficult to distinguish from GBS, but can be diagnosed if there is deterioration after 8 weeks from onset or more than three further deteriorations during disease course.33 With the notable exception of diphtheritic neuropathy, which is demyelinating-type and discussed later, most other acute peripheral neuropathies are of axonal type. Numerous infectious agents can directly damage peripheral nerves (eg, HIV) and some have the additional ability to trigger GBS (eg, herpes simplex virus). Tick paralysis should be considered if endemic.34 A flu-like prodrome lasting 5–10 days may be followed by rapidly progressive symmetric ascending flaccid paralysis over 2–6 days. In Lyme disease,35 painful, often asymmetric polyradiculoneuropathy may occur several months after initial tick bite or after a characteristic erythema migrans rash. Isolated or bilateral facial weakness has also been reported. CSF shows raised lymphocytes and protein, and there is serological evidence of Lyme disease. Consumption of certain plants (eg, buckthorn)28 or intoxication with lead, arsenic36 or thallium have all caused a rapidly progressive polyneuropathy mimicking GBS. Raw puffer fish (fugu) is a Japanese delicacy, but if prepared incorrectly may result in tetrodotoxin poisoning and rapidly progressive paralysis within hours of consumption.37 Currently, there is no antidote and treatment is supportive. Porphyric neuropathy38 is rare but shares several clinical features with GBS, although usually more asymmetric. The onset can be acute and progression to nadir occurs in 4 weeks. CSF also shows albuminocytological dissociation, and nerve conduction studies show an axonal-type neuropathy. In half the cases, the weakness starts in the upper limbs and may therefore be confused with the pharyngeal-cervical-brachial variant. Invariably, patients report severe abdominal pain and show psychiatric symptoms or have seizures before developing porphyric neuropathy. The diagnosis relies upon showing urinary porphyrins under ultraviolet light or following exposure to oxygen.
Neuromuscular junction disorders
Autoimmune diseases (eg, myasthenia gravis or Lambert–Eaton myasthenic syndrome), exposure to certain toxins (eg, botulinum toxin), consumption of various plants (eg, hemlock) or indeed animal bites (eg, cobra or krait) result in neuromuscular dysfunction and, in some cases, rapidly progressive paralysis in the absence of sensory disturbance. In myasthenia gravis, there is usually also demonstrable muscle fatigability.
Neuromuscular weakness related to critical illness
Neurologists are frequently asked to assess patients on the intensive care unit who develop generalised weakness or are having difficulty weaning from mechanical ventilation. Although GBS may develop in this setting, neuromuscular weakness related to critical illness should always be considered first.39 Clinical assessment may be difficult as patients are often encephalopathic. Those receiving high-dose intravenous glucocorticoids (eg, for status asthmaticus) are at particular risk of developing critical illness myopathy, which typically affects proximal more than distal muscles and starts several days after treatment initiation. There is sometimes facial weakness although the ocular muscles are usually spared. The serum creatine kinase is raised and nerve conduction studies may show slight motor nerve abnormalities. Patients with severe sepsis may develop critical illness neuropathy, which has a worse prognosis. Weakness is often more distal and associated with sensory disturbance. Facial weakness is less common. The serum creatine kinase in these patients is usually normal and nerve conduction studies show a diffuse sensory motor axonal polyneuropathy.39 Many patients have both critical illness myopathy and neuropathy and these should be differentiated from cachectic myopathy, which may develop in patients following prolonged admission and is more slowly progressive. Prolonged use of neuromuscular blocking agents (eg, vecuronium bromide) also may lead to persistent weakness and failure to wean from the ventilator.
Muscle disorders
Acute myositis may develop following certain viral infections (eg, influenza A),40 and is associated with variable degree of rhabdomyolysis. Typically, weakness begins within 2 weeks of the start of flu-like symptoms. Patients often look unwell and report myalgia and fever. On examination, the muscles are tender to touch, while sensation is normal. Serum creatine kinase is greatly elevated (often >10 000 U/L) and liver transaminases are also deranged. Urine dipstick is positive for blood but there are no red blood cells on microscopy.
Acute hypokalaemic periodic paralysis may mimic GBS.41 Two-thirds of cases are due to the familial form, which is autosomal dominant, and typically affects Caucasians. The paralysis often follows a high carbohydrate meal or a prolonged period of exertion. Thyrotoxic periodic paralysis is more prevalent among Asian, Hispanic and Native American males, and may be triggered by excess endogenous or exogenous thyroid hormones.
Functional illness
It is not uncommon to see patients presenting to the emergency room with vague sensory symptoms or odd patterns of weakness, which can be difficult to explain.42 In some patients, this may represent the early stages of GBS, but instead are dismissed as functional, especially if the deep tendon reflexes are present. By contrast, conversion disorder may be mistaken for GBS, although patients rarely display the core clinical features as described above.
Conditions mimicking MFS, Bickerstaff's brainstem encephalitis and the pharyngeal-cervical-brachial variant of GBS
Although MFS, Bickerstaff's brainstem encephalitis and the pharyngeal-cervical-brachial variant of GBS are rare, they should always be considered in patients presenting to the emergency room with symptoms and signs suggesting progressive brainstem dysfunction. Other neurological conditions, which commonly mimic these GBS variants include: brainstem stroke, myasthenia gravis, botulism, infective or inflammatory rhombencephalitis and bacterial, carcinomatous or lymphomatous meningitis. Wernicke's encephalopathy is the most important differential for patients with suspected Bickerstaff's brainstem encephalitis (box 3).
Differential diagnoses for Miller Fisher syndrome, Bickerstaff's brainstem encephalitis and pharyngeal-cervical-brachial weakness
-
Myasthenia gravis
-
Brainstem stroke (eg, basilar artery occlusion)
-
Diphtheritic neuropathy
-
Botulism
-
Rhombencephalitis
-
Infective (eg, listeriosis, tuberculosis, brucellosis, Lyme disease, herpes simplex virus, Epstein–Barr virus, JC virus, toxoplasmosis, cryptococcosis)
-
Autoimmune (eg, multiple sclerosis, sarcoidosis, Behçet's disease, systemic lupus erythematosus
-
Malignancy (eg, lymphoma, paraneoplastic syndrome)
-
-
Basal meningitis (Inflammatory, infective, carcinomatous and lymphomatous)
Brainstem stroke
Posterior circulation or brainstem strokes produce a variety of well-described syndromes determined by specific vascular territories. Some patients, for example, those with vertebral artery dissection, may not have risk factors for stroke, but instead may report recent neck injury (eg, whiplash) and present with neck pain. Typically, brainstem strokes are unilateral (eg, lateral medullary syndrome) unless the basilar artery is affected, in which case the ischaemic lesion often localises to the mid-pons, where it extends from the paramedian pontine base to the tegmentum. In most patients, the symptoms are progressive and many report vertigo, nausea and headache, two or more weeks beforehand.43 Limb weakness is usually asymmetric and often unilateral at onset. Facial and bulbar weakness and lower limb ataxia are also common. Patients nearly always show eye signs: typically horizontal gaze paresis or internuclear ophthalmoplegia. Pontine lesions produce pinpoint pupils. Some patients have pseudobulbar lability. ‘Top of basilar’ strokes produce a different syndrome, with ischaemic lesions in the midbrain, thalami, temporal and occipital lobes. The underlying cause is usually embolic. Sensation and weakness often remain intact, but patients have problems with vertical gaze and convergence. The pupils are enlarged and react slowly or incompletely. Altered mentation, hallucinations and memory problems may develop, depending on the area of ischaemia.
Myasthenia gravis
Myasthenia gravis is characterised by fluctuating weakness in voluntary muscles, which is made worse by exertion and often precipitated by certain drugs (eg, β-blockers) or systemic illness (eg, sepsis).44 Careful history taking often shows that symptoms have been present for some time. The presentation is highly variable, but there are four subtypes: generalised 50%, ocular 13%, ocular and bulbar 17%, and ocular and limbs 20%. Commonly (85%), patients present with fluctuating ptosis or diplopia. Ptosis is often asymmetric and increases with prolonged upgaze and may resolve with the ‘ice-pack’ test. The pupils are normal. Ophthalmoparesis often fluctuates from one examination to the next, whereas in MFS or Bickerstaff's brainstem encephalitis, ophthalmoparesis is progressive. Lateral rectus palsy is most frequent and therefore patients often report horizontal diplopia. Facial weakness may also occur and weakness of eye closure (orbicularis oculi) may occur in isolation. Fifteen per cent of patients present with bulbar symptoms, which may be obvious or subtle; for example, with a history of frequent chest infections secondary to silent aspiration. Some patients present with head droop, which may occur in the pharyngeal-cervical-brachial variant of GBS. Similarly, in myasthenia gravis, limb weakness is also usually proximal and more obvious in the upper limbs. The diagnosis of myasthenia gravis is usually clinical but often supported by the presence of serum anti-acetylcholine receptor antibodies (85% in general myasthenia gravis, <50% ocular myasthenia) and showing decrement on low frequency (2–5 Hz) repetitive stimulation (75% generalised myasthenia gravis, <50% ocular myasthenia). Single-fibre electromyography is abnormal in >95% of patients but is non-specific. Anti-MuSK (muscle-specific kinase) antibodies occur in 40% of anti-acetylcholine receptor antibody-negative patients with myasthenia gravis; these characteristically present with facial, bulbar, neck, and respiratory muscle weakness with relative sparing of ocular muscles.
Diphtheritic neuropathy
Approximately 10% of patients with diphtheria develop neuropathy involving the cranial or peripheral nerves, which is demyelinating in nature and usually occurs within the first 2 months of infection. Fever is usually present and in the vast majority there is bulbar dysfunction early in disease course.45
Wernicke's encephalopathy
Wernicke's encephalopathy results from thiamine deficiency and is often associated with chronic alcoholism in the setting of poor nutrition or increased metabolic requirements (eg, systemic illness). Similar to Bickerstaff's brainstem encephalitis, patients typically present with differing degrees of encephalopathy, ocular dysfunction and ataxia.46 Rather than the hypersomnolence seen in Bickerstaff's brainstem encephalitis, patients are often profoundly disorientated and inattentive and appear indifferent to their surroundings. Gaze-evoked nystagmus is the most frequent ocular presentation. Although there may be ophthalmoparesis, especially in the horizontal plane, this is usually incomplete unless Wernicke's encephalopathy is very severe. Ptosis is rare, and the pupils are often sluggish or unreactive. Vestibular dysfunction without hearing loss is also common. Gait ataxia may be the first sign of Wernicke's encephalopathy and is often multifactorial in the alcoholic and, therefore, sometimes dismissed. Wernicke's encephalopathy is primarily a clinical diagnosis but supported by the presence of characteristic changes on MR brain imaging. These include symmetric T2-signal change around the third and fourth ventricles and the aqueduct and, in most cases, also the mamillary bodies. Laboratory demonstration of thiamine deficiency is not always needed for diagnosis, although thiamine status can be determined by measuring whole blood levels, which is more sensitive than measurement of serum levels or the plasma transketolase method. Parenteral thiamine must be given early and should not be delayed in suspected cases; it should be given before glucose to avoid deterioration.
Botulism
Intoxication with Clostridium botulinum toxin produces a pattern of weakness similar to MFS and the pharyngeal-cervical-brachial variant of GBS, and is increasingly seen in patients who inject black tar heroin. Interestingly, botulinum toxin type A, the most common cause of foodborne botulism, also binds to the gangliosides GQ1b and GT1a and may disrupt the same population of neurons.47 ,48 In addition to ptosis, ophthalmoplegia and faciobulbar weakness, the pupils are characteristically dilated and poorly reactive to light and accommodation. Descending paralysis in the absence of sensory disturbance may be complicated by respiratory failure, if severe. Some patients also develop autonomic symptoms, including dry mouth, postural hypotension and constipation. Foodborne botulism typically develops 12–72 h after ingesting contaminated food (eg, home-canned vegetables) and weakness may be preceded by nausea, vomiting and diarrhoea.49 Wound infections with C. botulinum spores, sometimes seen in drug users from contaminated supplies, and colonisation of the small intestine in newborns can also rarely occur. The diagnosis is confirmed with mouse bioassay, which involves intraperitoneal injection of the patient's serum, gastric secretions or stool into a mouse. The treatment is supportive and antitoxin should be administered early in suspected cases.
Rhombencephalitis
Brainstem encephalitis (rhombencephalitis) should be among the differential diagnosis in patients who develop rapidly progressive brainstem signs.50 When there is underlying infection, patients characteristically report headache, fever, nausea and vomiting, followed by brainstem, cerebellar and long tract signs. Respiratory depression and decreased consciousness are common and seizures may also occur. In some patients, meningeal symptoms are prominent and this narrows the differential further. Listeriosis is probably the most common cause, especially in the immunocompromised and among alcoholics or in healthy individuals who have consumed heavily contaminated food. It may also complicate the third trimester of pregnancy and lead to miscarriage. Its mortality is high and, therefore, ampicillin is often used empirically in patients at risk. Other infective causes depending on patients’ background should also be considered and include: tuberculosis, brucellosis, Lyme disease, herpes simplex virus, Epstein–Barr virus, JC virus, toxoplasmosis and cryptococcosis. A wide range of non-infective conditions may produce similar symptoms, although systemic illness, especially fever, are often less prominent.50 These include, but are not limited to: multiple sclerosis, sarcoidosis, Behçet's disease, systemic lupus erythematosus, lymphoma and paraneoplastic syndromes. Brainstem imaging is usually abnormal and there may be meningeal enhancement. CSF analysis, blood cultures and serological work-up are useful when abnormal or positive, but may be unrevealing, which can delay the final diagnosis.
Unusual presentations of GBS, MFS and their subtypes—Chameleons
Atypical forms of GBS and MFS can be difficult to diagnose, but should always be considered in the context of antecedent infection and monophasic disease course. Most of these variants are exceptionally rare, except for hyper-reflexic GBS.
Hyper-reflexic GBS
To many neurologists, GBS is a disorder that affects only peripheral nerves and, therefore, is frequently overlooked in the presence of normal or exaggerated deep tendon reflexes. However, two large studies2 ,3 indicated that 10% of patients with GBS had normal or brisk reflexes throughout the course of illness. Interestingly, these patients were more likely to present with pure motor weakness and, neurophysiologically, had features consistent with acute motor axonal neuropathy rather than acute inflammatory demyelinating polyradiculoneuropathy. Patients with hyper-reflexic GBS also often have anti-GM1 or anti-GD1a antibodies.2 The term ‘hyper-reflexic variant’ of GBS has been coined but we believe this is unnecessary. It is important to note that muscle tone is normal in patients with hyper-reflexic GBS. The exact localisation and mechanism which underpins hyper-reflexia in GBS remains unknown, but one theory is that antiganglioside antibodies may cross the blood–brain–barrier and disrupt intramedullary interneurones.2
Paraparetic GBS
There is another unusual localised variant of GBS6–8 in patients who develop isolated flaccid lower limb weakness without neurological findings in the upper limbs. In the cases described, bilateral ‘sciatic-like’ leg pain was prominent and appeared early, undoubtedly contributing to loss of leg function. Deep tendon reflexes were absent in the lower but not the upper limbs and supported localisation to the lumbar nerve roots. Moreover, CSF albuminocytological dissociation, normal routine laboratory findings and unremarkable MRI allowed differentiation from other causes of lumbar polyradiculopathy, including diabetes mellitus, vasculitis, infiltrative or compressive lesions and other infective agents. The diagnosis of so-called paraparetic GBS is supported by showing axonal-type neuropathy on nerve conduction studies, and the presence of serum antiganglioside antibodies.7
Bifacial weakness with paraesthesias
The bifacial weakness with paraesthesias variant of GBS9 should always be considered in patients who develop bilateral facial weakness in the absence of ophthalmoplegia or limb weakness. Most patients with the bifacial weakness with paraesthesias variant also report distal limb paraesthesias, elicited by careful sensory examination, allowing differentiation from other causes of bilateral facial weakness, including bilateral Bell's palsy, Lyme disease and sarcoidosis. The diagnosis of the bifacial weakness with paraesthesias variant is also supported by the presence of antecedent infection, CSF albuminocytological dissociation and evidence of peripheral nerve demyelination in the limbs.
Acute ataxic neuropathy
Two forms of incomplete MFS are characterised by profound ataxia in the absence of ophthalmoplegia.18 In the first, known as ataxic GBS, patients display ataxia in the absence of both ophthalmoplegia and Rombergism. In the second, known as acute sensory ataxic neuropathy, patients display ataxia in the absence of ophthalmoplegia but with Rombergism. To avoid nosological confusion, ataxic GBS and acute sensory neuropathy are grouped together and known collectively as acute ataxic neuropathy. Rather than being distinct entities, ataxic GBS and acute sensory ataxic GBS form a continuous spectrum, which is observed clinically and serologically. Patients with ataxic GBS are more likely to have antibodies against GQ1b, whereas monospecific IgG anti-GD1b antibodies without GQ1b reactivity are more common in patients with acute sensory ataxic neuropathy.18 The mechanism for cerebellar-like ataxia in these patients is thought to be from selective involvement of muscle spindle afferents by anti-GQ1b antibodies.51
Acute ophthalmoparesis/acute ptosis/acute mydriasis
There are rare incomplete forms of MFS in patients who develop isolated eye signs in association with anti-GQ1b antibodies, but without ataxia. In one study, 25% of 100 patients presenting with acute abducens palsy had anti-GQ1b antibodies.52 Patients who develop acute ophthalmoparesis19 may also develop facial or rarely bulbar weakness, reinforcing the concept that MFS is a spectrum of overlapping disorders. Ptosis20 and mydriasis21 are not cardinal features of MFS but may also appear in isolation and are associated with anti-GQ1b antibodies.
Conclusions
GBS and its only variant, MFS, have several subtypes, which present in various ways and can be difficult to diagnose at first. The recognition of these subtypes based on core clinical features (history of antecedent infection, monophasic disease course, and symmetrical cranial or limb weakness) often allows differentiation from other causes of acute flaccid paraparesis or brainstem syndromes. There are also more unusual localised forms of GBS and incomplete forms of MFS, which should always be considered, as the prognosis is often favourable. Early correct diagnosis—especially in GBS, MFS, Bickerstaff's brainstem encephalitis and the pharyngeal-cervical-brachial variant—is important and avoids unnecessary investigations and guides appropriate use of immunotherapy.
Key points
-
GBS and MFS subtypes form a continuous spectrum of discrete and overlapping syndromes.
-
GBS spectrum disorders should always be considered if relatively symmetric limb or cranial nerve weakness and ataxia, follow antecedent upper respiratory infectious symptoms or diarrhoea. This is supported by the presence of distal paraethesias, CSF albuminocytological dissociation, abnormal nerve conduction studies, or anti-ganglioside antibodies.
-
GBS is very likely in patients who develop acute flaccid paralysis with facial weakness.
-
MFS and the pharyngeal-cervical-brachial variant of GBS are frequently mistaken for myasthenia gravis, botulism or brainstem stroke.
-
10% of patients with GBS have normal or exaggerated deep tendon reflexes.
References
Footnotes
-
Contributors BRW researched data for the article and drafted the article. NY revised the article.
-
Funding Supported by Singapore National Medical Research Council (IRG 10nov086 and CSA/047/2012 to NY).
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by John Winer, Birmingham, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Ataxic Guillain–Barré syndrome and acute sensory ataxic neuropathy form a continuous spectrum
- Anti-GQ1b IgG antibody syndrome: clinical and immunological range
- Stumbling towards a diagnosis
- Pharyngeal-cervical-brachial variant of Guillain–Barré syndrome
- Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome
- Treatment dilemmas in Guillain-Barré syndrome
- Miller Fisher syndrome
- Blood–brain barrier destruction determines Fisher/Bickerstaff clinical phenotypes: an in vitro study
- Nationwide survey of patients in Japan with Bickerstaff brainstem encephalitis: epidemiological and clinical characteristics
- Bickerstaff's encephalitis