Article Text

Abstract
Mitochondrial diseases are inherited disorders of oxidative phosphorylation that present with a multitude of clinical features in different combinations and with various inheritance patterns. To complicate the issue further, the clinical features of mitochondrial disorders overlap with common neurological and non-neurological diseases. This presents a diagnostic challenge: when is a rare mitochondrial disease responsible for a more ‘common or garden’ neurological presentation, and how often are neurologists missing them in routine clinical practice? Here, we briefly review some common clinical features associated with mitochondrial disease, and provide some clues as to how patients with these mitochondrial disorders might be identified. We discuss both ‘chameleons’—mitochondrial disorders that may look like something else, and ‘mimics’—other conditions that may clinically resemble mitochondrial disease. The diagnosis sometimes needs highly specialised tests, but the advent of ‘next generation’ sequencing will simplify the clinical approach over the next few years.
- MITOCHONDRIAL DISORDERS
Statistics from Altmetric.com
Introduction
Mitochondrial disease may present with a multitude of clinical features in different combinations. The most energy-dependent organs of the body, such as the brain, heart, skeletal muscles and endocrine pancreas, are commonly affected by mitochondrial disease as they are most vulnerable to the dysfunction of oxidative ATP production.1 ,2 Different genetic causes of mitochondrial disease may lead to very similar phenotypes, but on the other hand, very different clinical presentations can arise from the same underlying genetic cause. Most patients do not show the ‘full house’ of features typical of the canonical mitochondrial clinical syndromes, and unfortunately the clinical and genetic spectrum evolves on a yearly basis. Mitochondrial disorders affect fewer than 1 in 5000 adults,3 so there is only a low likelihood of seeing a new case in a general neurology clinic. How can a neurologist hope to spot these rare patients in routine practice? This overview of the major mitochondrial ‘chameleons’ and ‘mimics’ will provide a guide (box 1).
Mitochondrial mimics—a non-comprehensive list
Neuropathy
Charcot–Marie–Tooth disease: in particular axonal forms, but also demyelinating and intermediate forms, have been described in mitochondrial disease.
Epilepsy
Epilepsy in mitochondrial disease is usually accompanied by additional clinical features or is part of an epileptic encephalopathy. In the acute de novo setting, it is important to exclude encephalitis. Other forms of epileptic encephalopathy should be considered in the subacute setting, particularly if refractory to treatment (eg, epilepsy syndromes such as SCN4A gene-related, or other metabolic disorders such as glutaric aciduria type I).
Other causes of myoclonic epilepsy with/without ataxia include ceroid lipofuscinosis and gluten encephalopathy.
External ophthalmoplegia
Myasthenia gravis and oculopharyngeal muscular dystrophy
Myopathy
Isolated mitochondrial myopathy is rare, but resembles genetic and acquired limb-girdle syndromes and Dok-7 congenital myasthenia.
Stroke-like episodes (and extensive leukoaraiosis)
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
Other leukodystrophies
Ataxia
Fragile X-associated tremor/ataxia syndrome (FXTAS)
Spinocerebellar ataxias including Friedreich's ataxia and vitamin E deficiency
Optic atrophy and pigmentary retinopathy
Pigmentary retinopathy (due to other causes)
Susac's syndrome (retinopathy, encephalopathy and deafness)
Usher's syndrome (retinitis pigmentosa with deafness)
Wolfram's syndrome (optic atrophy with deafness and/or diabetes)
Conventionally, mitochondrial disease refers to inherited disorders that are characterised by dysfunction of mitochondrial oxidative phosphorylation. These can be due to mutations in both mitochondrial DNA (mtDNA) and nuclear DNA, and can present with autosomal dominant, autosomal recessive, X-linked or mitochondrial (ie, maternal) inheritance patterns (box 2). Genetic defects that result in mitochondrial disease may also arise de novo in the maternal germline, leading to an apparently sporadic disease, so even for these genetic disorders the family history is not a reliable a guide. Here we briefly review some common clinical conditions and phenotypes associated with mitochondrial disease, and provide some clues as to how to recognise patients with these mitochondrial conditions.
Basic primer on mitochondrial biology and genetics
Mitochondria are intracellular organelles that form a dynamic network and are present in all eukaryotic cells (figure 6).
The essential role of mitochondria is the production of ATP to meet the cellular energy demands.
Mitochondria are also involved in other functions, such as the control of cellular calcium levels, fatty acid oxidation and apoptosis.
Mitochondria contain their own DNA (mtDNA), a circular, double-stranded molecule 16,569 base-pairs in length; mtDNA is inherited maternally.
MtDNA contains 37 genes, of which two are rRNA genes, 22 are tRNA genes, and the remaining 13 code for components of the mitochondrial respiratory chain complexes.
Mitochondria depend upon their cellular surroundings and the nuclear DNA, as most of the protein constituents of the respiratory chain and the several factors needed for mtDNA maintenance, translation and respiratory chain assembly are nuclear-encoded.
Mitochondrial diseases can therefore be caused by mutations of mtDNA or nuclear DNA, and in some instances the nuclear DNA defect leads to secondary mtDNA damage (so-called mtDNA maintenance disorders).
Neuropathy
In addition to the central nervous system, mitochondrial disorders may also affect the peripheral nerves. A common presentation is a predominantly axonal, length-dependent, distal neuropathy, for example, in neuropathy, ataxia and retinitis pigmentosa (NARP) syndrome.4 As with other classic mitochondrial clinical syndromes, many patients have only one or two of the canonical features, and some present with a neuropathy resembling Charcot–Marie–Tooth disease.5 In mitochondrial disease due to pathogenic mutations in the nuclear gene POLG, encoding the mitochondrial DNA polymerase γ, severe axonal sensory neuropathy or a dorsal root ganglionopathy can cause debilitating sensory ataxia, resembling an inflammatory or autoimmune disorder (particularly Sjögren's syndrome).6
Although not conventionally regarded as a mitochondrial disease, mutations in MFN2 encoding a mitochondrial dynamics protein mitofusin 2, result in Charcot–Marie–Tooth disease type 2A, a form of axonal neuropathy with a dominant inheritance pattern.7 The clinical picture of MFN2-associated disease can be more complex, with reports of axonal neuropathy with optic atrophy, or with associated cognitive impairment, corticospinal tract involvement and sensorineural hearing loss. Mutations in MFN2 may also give rise to an optic atrophy ‘plus’ phenotype with multiple mtDNA deletions.8
In patients with axonal neuropathy, additional clinical features such as progressive external ophthalmoplegia (PEO), retinitis pigmentosa or ataxia might prompt further investigations to identify an underlying mitochondrial cause of the neuropathy. Occasional patients with mitochondrial disease have a demyelinating neuropathy.9 ,10
Epilepsy
Epilepsy is a common feature in mitochondrial disease. Patients with the syndrome of myoclonic epilepsy with ragged-red fibres (MERRF), associated with the mtDNA m.8344A>G point mutation in the mitochondrial tRNA(Lys) gene MTTK, develop myoclonus and generalised seizures.11 There are often additional features such as ataxia, myopathy, hearing loss, ptosis and multiple lipomatosis.11
Epilepsy is also common in patients with mitochondrial disease due to POLG mutations.12 The mean age of presentation of POLG epilepsy is around 18 years; frequent accompanying symptoms are migraine-like headache, ataxia and neuropathy. Epilepsy associated with POLG mutations often has an occipital predilection, causes intractable epilepsia partialis continua, and may progress insidiously to severe, life-threatening generalised status epilepticus.13 It is very important to detect epilepsy due to POLG mutations promptly, as there is a major risk of progression to treatment-resistant status epilepticus. Sodium valproate may cause life-threatening liver failure in patients with POLG mutations and for this reason must be avoided.12 ,13
Distinguishing mitochondrial epilepsy from non-mitochondrial epilepsy can be challenging, especially in the early stages. However, most patients with mitochondrial epilepsy have additional features either in the family, or on clinical investigation.
PEO and myopathy
PEO is a common manifestation of mitochondrial disease (figure 1). This clinical entity is defined as the combination of a bilateral—but not necessarily symmetrical—ptosis and a progressive limitation of eye movements (external ophthalmoplegia). Some patients present with ptosis only, without apparent restriction of eye movements. Diplopia is uncommon in PEO, and if present, it is usually transient. PEO strongly suggests a mitochondrial disease. Patients with ‘PEO-plus’ have involvement of other organs as well.14
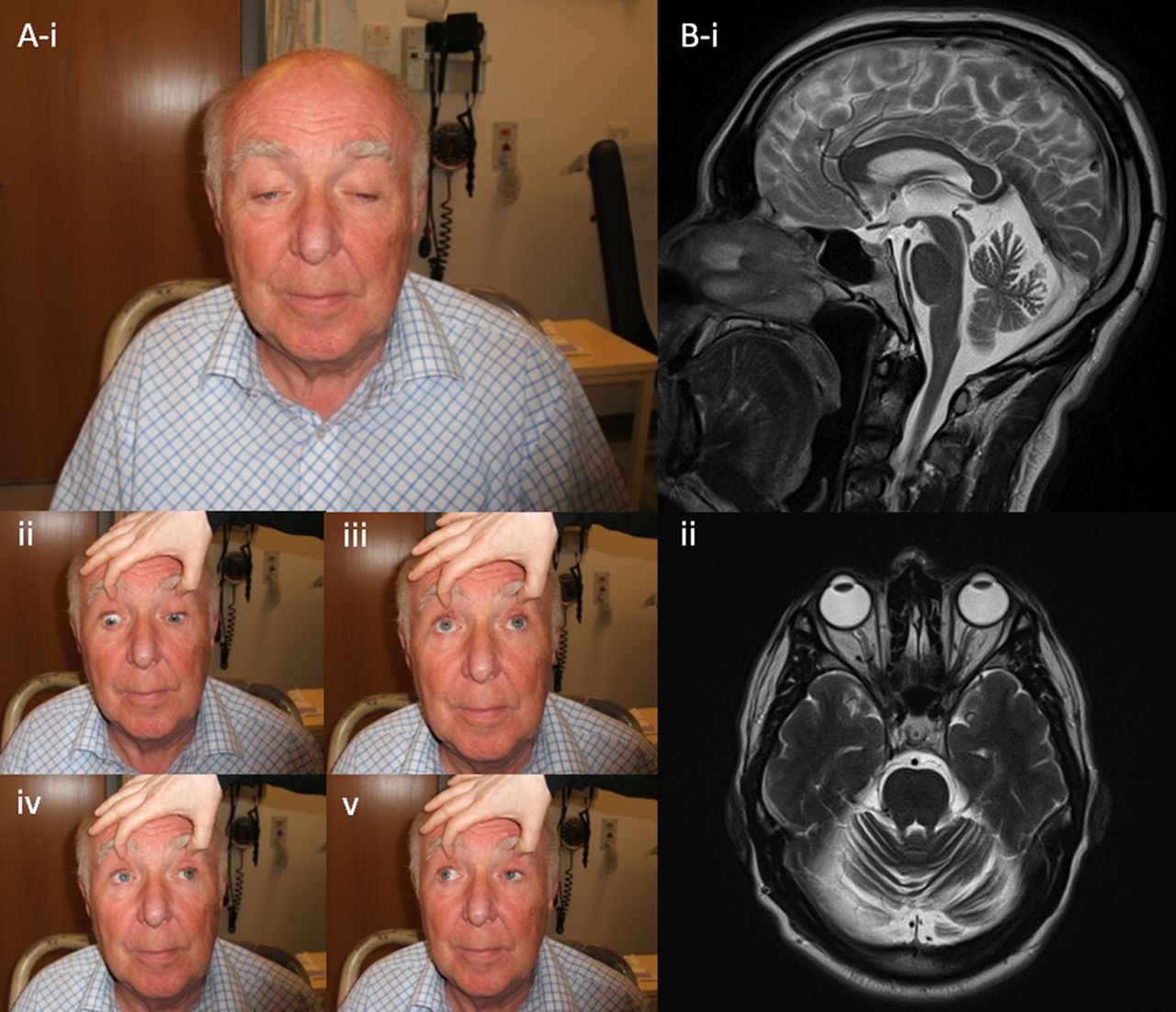
Patient with progressive external ophthalmoplegia due to mutations in the SPG7 gene. (A) Note the marked, slightly asymmetric ptosis and the restriction of eye movements, particularly in the upward direction (iii). (B) T2-weighted brain MRI demonstrates diffuse cerebellar atrophy. (Figure originally published in Pfeffer et al, Brain 2014.31)
Molecular causes of PEO include mtDNA rearrangements, such as large-scale deletions15 and mutations in mitochondrial tRNA genes. In addition, several nuclear gene defects may result in multiple mtDNA deletions, giving rise to PEO with either an autosomal dominant or recessive inheritance pattern: PEO1, POLG and RRM2B are common examples.16–18 A molecular diagnosis is important because of strikingly different recurrence risks, and a different spectrum of complications requiring disease surveillance.
In Kearns-Sayre syndrome, PEO is accompanied by pigmentary retinopathy and at least one other feature (myopathy, heart block, cerebellar ataxia or high cerebrospinal fluid protein), although many patients with the syndrome have additional clinical features (eg, pyramidal signs, short stature or diabetes mellitus). By definition, Kearns-Sayre syndrome symptoms appear before 20 years of age.19
Mitochondrial PEO should be distinguished from other causes of ptosis or generally decreased eye movement. For example, the absence of prominent fatigability makes ocular myasthenia gravis less likely, while fluctuating diplopia makes ocular myasthenia more likely. It is worth remembering that acetylcholine receptor antibodies are often absent in ocular myasthenia. Longstanding dysphagia (predating the PEO) makes oculopharyngeal muscular dystrophy more likely, confirmed in most cases by a mutation in PABPN1. The ptosis and blepharospasm characterising myotonic dystrophy type 1 are usually accompanied by obvious clinical myotonia; the overall facial weakness is usually more pronounced than the ptosis.20 Other myopathies, such as nemaline myopathy and desminopathy, may show ptosis without PEO. Normal thyroid function tests make Graves’ orbitopathy unlikely.
Proximal or generalised myopathy is also fairly common in mitochondrial disease but is usually associated with other features. In mitochondrial disease, the serum creatine kinase is often normal, as is the electromyogram. On muscle biopsy, the classic findings are first, the presence of excessive cytochrome c oxidase (COX)-deficient fibres on the combined COX and succinate dehydrogenase stain, and second, ragged-red fibres using the Gomori trichrome stain. However, these features also develop as part as healthy aging, and must be interpreted with caution. Molecular genetic investigations on DNA isolated from a muscle biopsy specimen may show mtDNA abnormalities, such as point mutations or rearrangements (large-scale deletion, duplication). In the case of an underlying nuclear gene mutation affecting mtDNA maintenance, there may be multiple secondary mtDNA deletions or mtDNA depletion.
Inclusion body myositis is known to show COX-negative fibres and mtDNA deletions in muscle.21 Polymyositis and dermatomyositis may cause similar changes, usually interpreted as secondary, but their precise mechanism is unclear.
Stroke-like episodes
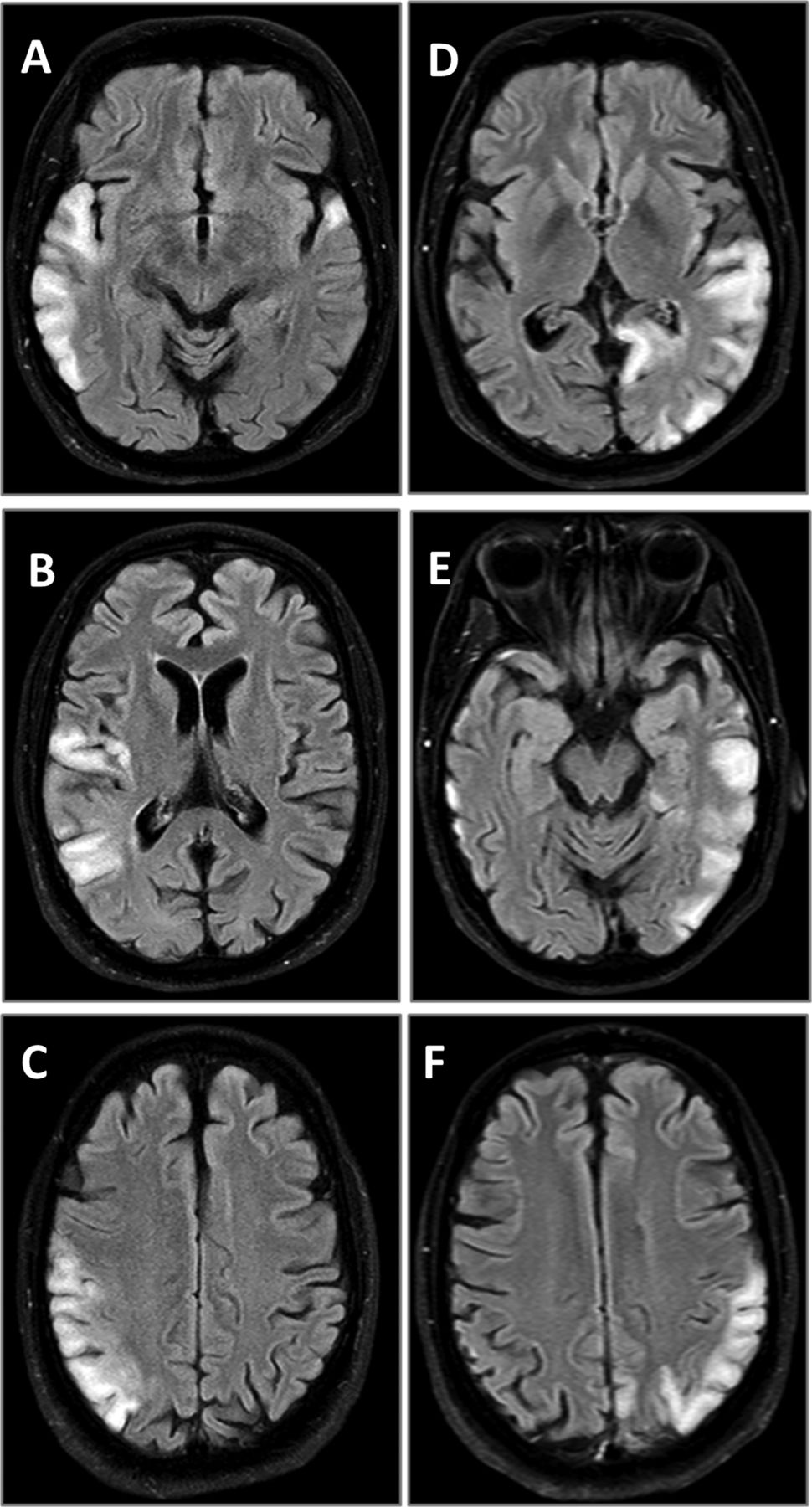
The stroke-like episodes in mitochondrial disease have been classically associated with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome. The radiological appearance of stroke-like lesions associated with MELAS may resemble that of common ischaemic strokes, although the stroke-like lesions do not conform to vascular territories, may change size and even location in time, and later may completely disappear (figure 2). The occipital and parietal regions of the brain seem to be particularly vulnerable.22 ,23 The stroke-like episodes have been considered the classic clinical hallmark of MELAS syndrome, but also occur in patients with autosomal recessive POLG mutations. In both, the occipital regions of the brain are frequently affected, and patients often show clinical signs suggesting occipital brain dysfunction, such as homonymous hemianopia or cortical blindness.24 Brain imaging studies show that the stroke-like lesions may change size and location with time (figure 2).23 Their likely underlying mechanism is a metabolic deficiency due to the impaired oxidative energy production, although their exact cause is unclear.

Serial MRI (fluid attenuation inversion recovery sequence) of two stroke-like episodes in a patient with the m.3243A>G mutation. (A–C) The first episode; (D–F) the second episode. Note that the hyperintense changes in temporal, parietal and occipital lobes do not conform to the vascular territories.
When compared to common ischaemic stroke, a mitochondrial stroke-like episode typically follows a more insidious time course. Migraine-like headache, visual field loss and flashing visual disturbances often accompany or precede any motor and sensory deficits. Seizures are common in stroke-like episodes. The radiological findings associated with stroke-like episodes change over time, and repeated brain imaging may help in diagnosis. Similar MRI findings in the posterior part of the brain, and particularly the thalamus, also commonly occur in POLG-associated mitochondrial disease.13
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common heritable cause of both stroke and vascular dementia in the adult population.25 Migraine with aura is very common in CADASIL, as is cognitive decline and dementia in advanced disease. Brain MRI typically shows an extensive pattern of white-matter ischaemic lesions; in CADASIL, the temporal lobes are commonly affected, which is atypical for other causes of ischaemic lesions. Molecular genetic testing for pathogenic mutations in the NOTCH3 gene is the gold standard for the diagnosis of CADASIL.25
Primary central nervous system vasculitis presents at the median age of 50 years with headache, altered cognition and stroke-like symptoms.26 Cerebrospinal fluid investigation typically (in 80–90% of cases) shows a greatly raised protein concentration; in contrast, an elevated cerebrospinal fluid lactate concentration might be more suggestive of a mitochondrial disorder. Multiple, bilateral cortical and subcortical infarctions are common in MR brain imaging in CADASIL. Cerebral angiography (either conventional digital subtraction or MR angiography) shows alternating segments of stenosis and arterial occlusions.
Ataxia
Ataxia is common in mitochondrial disease.27 ,28 Cerebellar ataxia may develop in patients with the m.3243A>G, m.8344A>G and m.8993T>G/C mtDNA mutations (originally described in association with clinical MELAS, MERRF and NARP syndromes, respectively). The m.8993T>G/C mutations are often encountered in Leigh's syndrome, but may also occur in adult-onset NARP syndrome, characterised by the combination of peripheral neuropathy and ataxia.4 ,29 Among nuclear gene defects leading to mitochondrial disease, POLG mutations are frequently associated with both cerebellar and sensory ataxia.6 ,30 More recently, spastic ataxia has been described as part of the phenotype related to pathogenic SPG7 mutations that lead to disordered mtDNA maintenance.31
‘Pure’ or isolated cerebellar ataxia is unusual in mitochondrial disease. It is therefore important to exclude common toxic, metabolic, inflammatory and degenerative causes before reaching a mitochondrial diagnosis. Nonetheless, classic mtDNA mutations can cause a disorder resembling typical inherited spinocerebellar syndromes and Friedreich's ataxia.32
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a disorder resulting from moderate expansions (55–200) of a CGG repeat in the FMR1 gene, in which expansions of more than 200 repeats result in fragile X syndrome.33 Clinical features of FXTAS include action tremor and cerebellar gait ataxia, sometimes with parkinsonism, neuropathy, dysautonomia and executive dysfunction.33 The clinical resemblance of FXTAS to mitochondrial disorders may be explained by the disruption of mitochondrial protein import and processing that has been reported in this condition.34 ,35
Optic neuropathy and pigmentary retinopathy
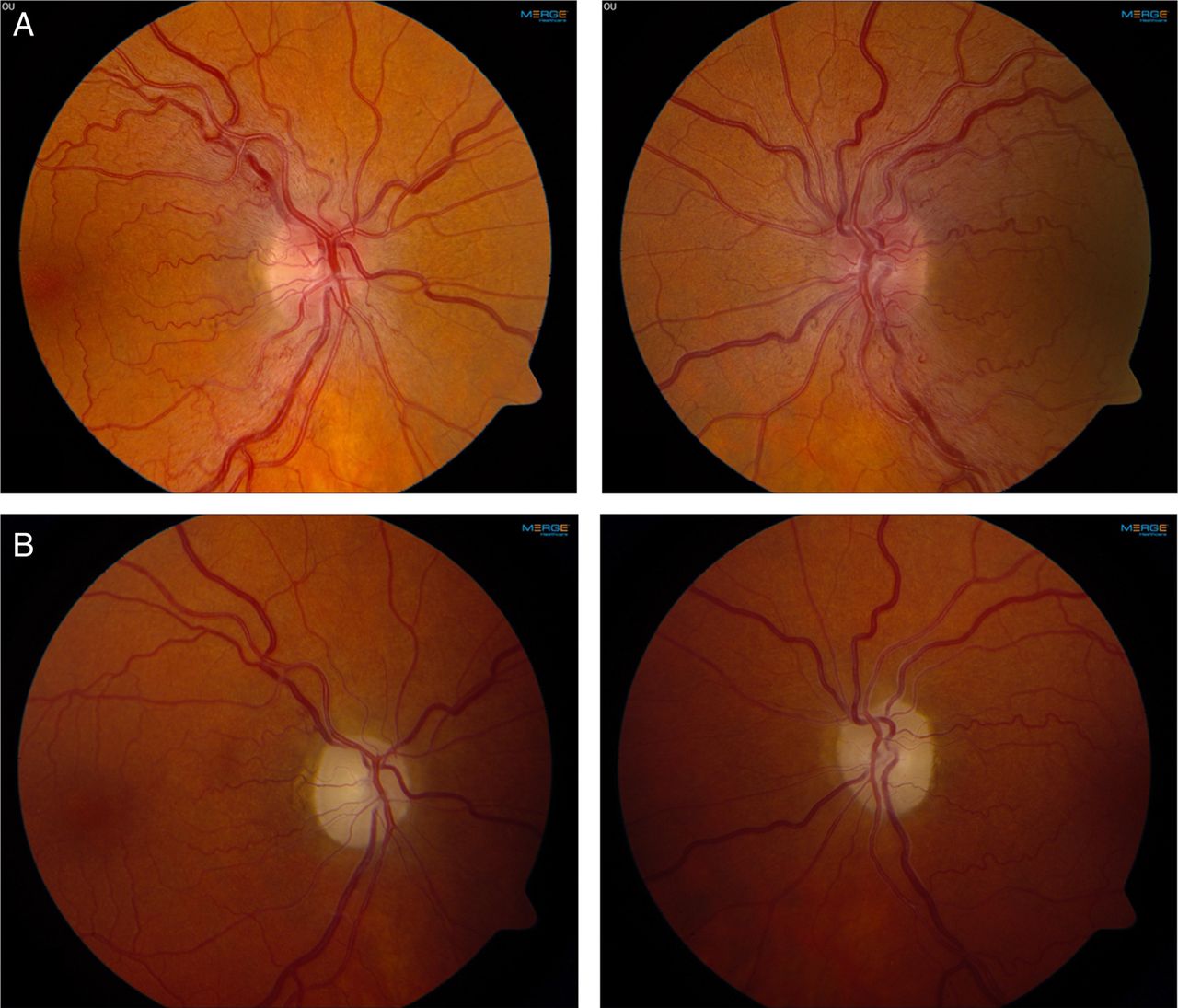
The most common mitochondrial disorder that results in optic neuropathy is Leber's hereditary optic neuropathy (LHON). This is caused, in almost all cases, by one of three mtDNA mutations, each encoding for a respiratory chain (RC) complex 1 subunit. Three point mutations that encode RC complex I subunits—m.11778G>A, m.14484T>C and m.3460G>A—are found in the vast majority of patients with LHON.36 Clinically, it is characterised by bilateral, subacute visual failure due to degeneration of the retinal ganglion cells and the optic nerves.37 The loss of vision typically occurs in young adulthood, and in the acute phase there may be increased vascularity of the optic nerve head with tortuous vessels, creating the impression of optic disc swelling (figure 3). In one reported patient, late-onset LHON mimicked Susac's syndrome, a rare autoimmune disorder resulting in microvascular occlusions in the brain, retina and inner ear.38 Some patients with LHON also present with clinical and MRI features suggesting multiple sclerosis. The disorder resulting from the co-occurrence of LHON and multiple sclerosis (‘Harding's disease’) has a distinct phenotype, suggesting a mechanistic interaction between these two diseases39 (figure 4). OPA1 encodes a dynamin-related GTPase-like OPA1 protein that is involved in mitochondrial dynamics. 3OPA1 were first described in optic atrophy type 1, a dominantly inherited condition resulting in progressive loss of visual acuity.40 Further studies have shown that OPA1 also gives rise to more complex phenotypes (‘optic atrophy plus’) with multiple mtDNA deletions.41 In the context of mitochondrial disease, pigmentary retinopathy is most typically associated with Kearns–Sayre and NARP syndromes.4 ,19
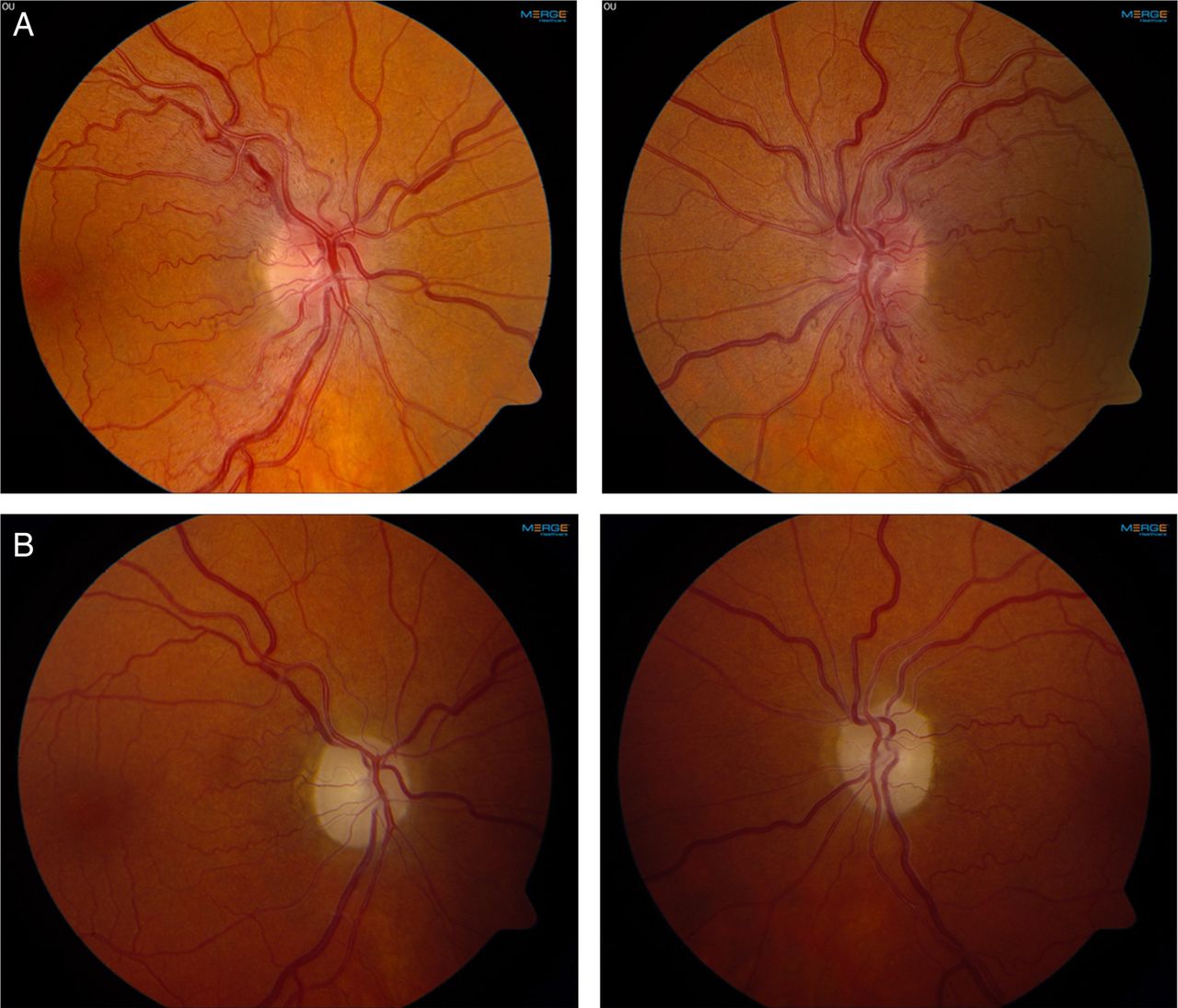
Leber hereditary optic neuropathy due to the 11778G>A mutation in mitochondrial DNA. (A) Optic disc swelling in the acute phase of visual loss. (B) Optic atrophy and pale optic discs in the chronic phase of the disease. Photos courtesy of Mr Patrick Yu-Wai-Man.
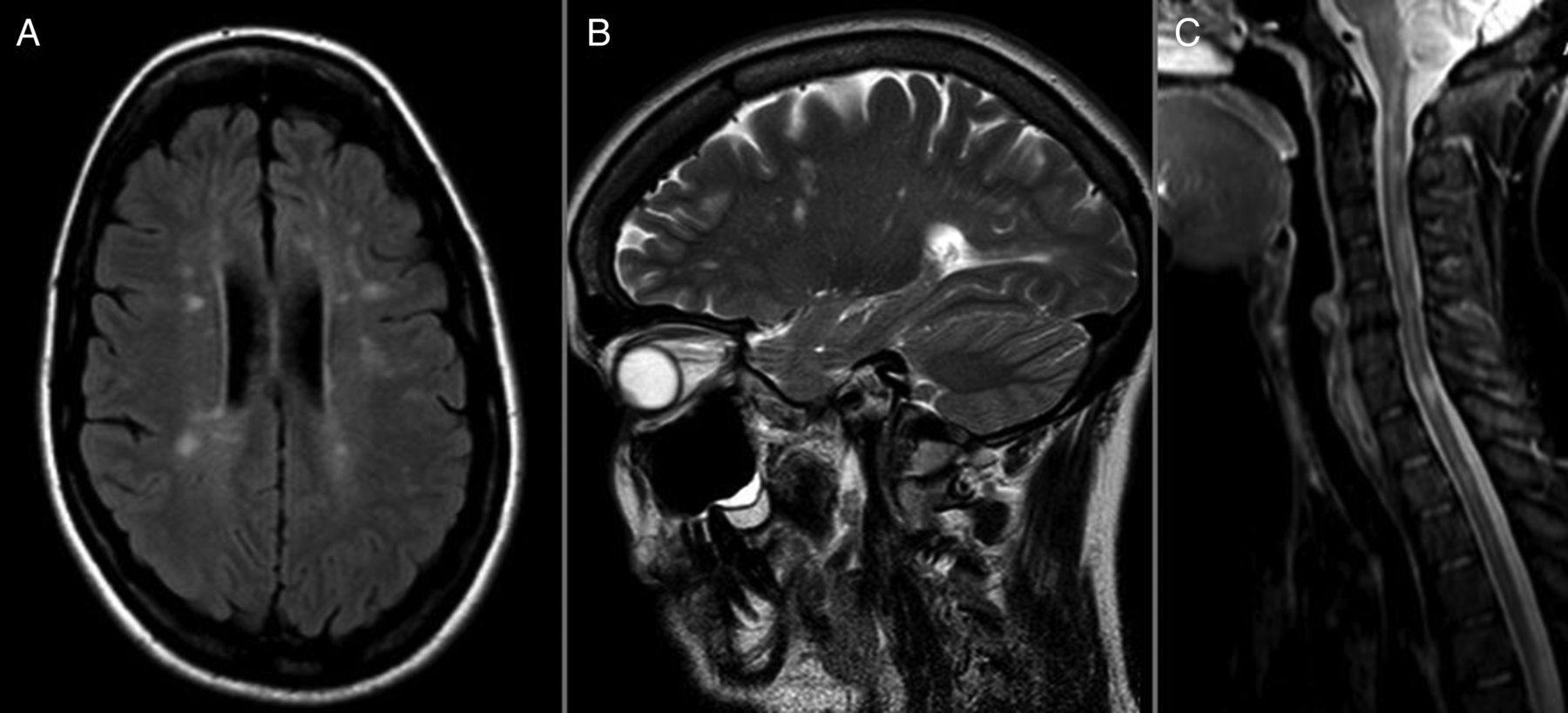
MRI findings in a patient with co-occurring multiple sclerosis and Leber hereditary optic neuropathy due to the 11778G>A mutation in mitochondrial DNA. (A) Fluid attenuation inversion recovery sequence. Multiple periventricular and juxtacortical high-signal white matter lesions. (B) T2-weighted sequence. Dawson finger-resembling lesions in sagittal orientation. (C) T2-weighted sequence. Multiple high signal lesions in the cervical cord. (Figure originally published in Pfeffer et al, Neurology 2013.39)
Although LHON is a strong possibility in patients with subacute painless visual failure, it is important to consider structural, inflammatory and other genetic causes and investigate accordingly. LHON preferentially affects young adult men, but can occur in children and the elderly, and presents in an identical way in women. Having excluded structural and inflammatory causes, other genetic causes include Wolfram's syndrome, which may present with a bilateral optic neuropathy. Wolfram's syndrome 1 is an autosomal recessive condition caused by mutations in the WFS1 gene. It is clinically defined by the combination of diabetes insipidus, diabetes mellitus, optic atrophy and deafness, but incomplete penetrance is much more common than previously thought.42 Mutations in CISD2 can result in Wolfram's syndrome 2 that clinically resembles Wolfram's syndrome 1 except for the absence of diabetes insipidus.
If there is a pigmentary retinopathy, clinicians should consider Usher's syndrome. This is the most common cause of inherited deafness–blindness in humans.43 The inheritance pattern is autosomal recessive. Usher's syndrome is genetically heterogeneous, with 12 loci and nine causative genes identified thus far. There are three subtypes (USH 1–3), defined based on the severity of hearing impairment, the presence of vestibular dysfunction and the age of onset of retinitis pigmentosa.43
Movement disorders
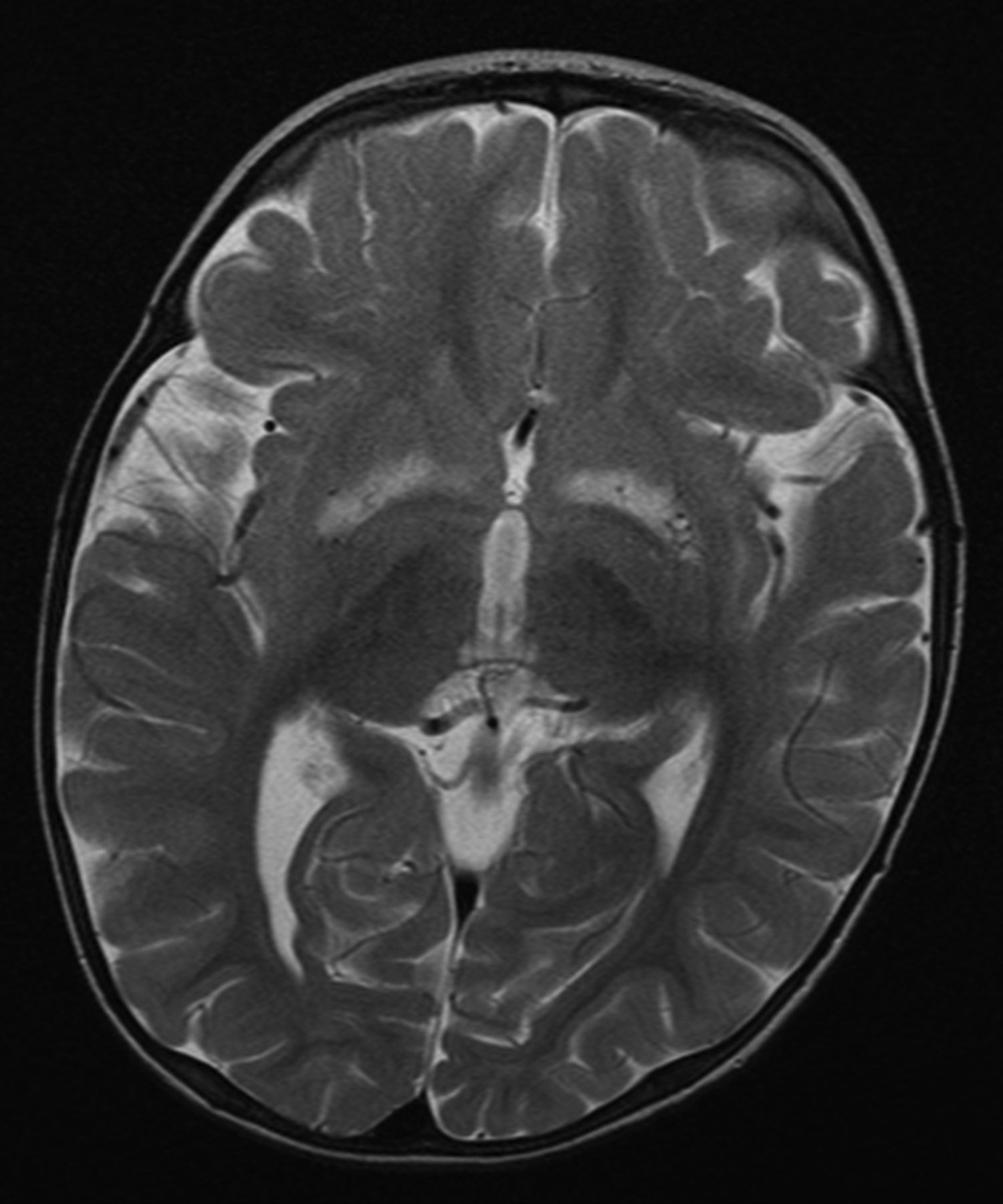
Patients with mitochondrial disease may develop various types of movement disorders, such as parkinsonism, dystonia and myoclonus.44 Parkinsonism may develop in association with POLG mutations,45 but also with mtDNA mutations such as those associated with LHON,46 the m.8344A>G mutation47 and the large-scale ‘common’ mtDNA deletion.48 In the context of mitochondrial disease, dystonia may develop in association with Leigh's syndrome, the m.3243A>G mutation and the LHON mutations.49–51 Myoclonus is most commonly associated with MERRF and the m.8344A>G mutation, although myoclonus may present with several other mitochondrial disorders, such as those due to POLG mutations.11 ,52 Chorea develops only rarely in mitochondrial disease.53 ,54 In some cases, the characteristic finding of bilateral, symmetric putaminal necrosis on brain MRI may help recognition of a mitochondrial movement disorder (figure 5).51
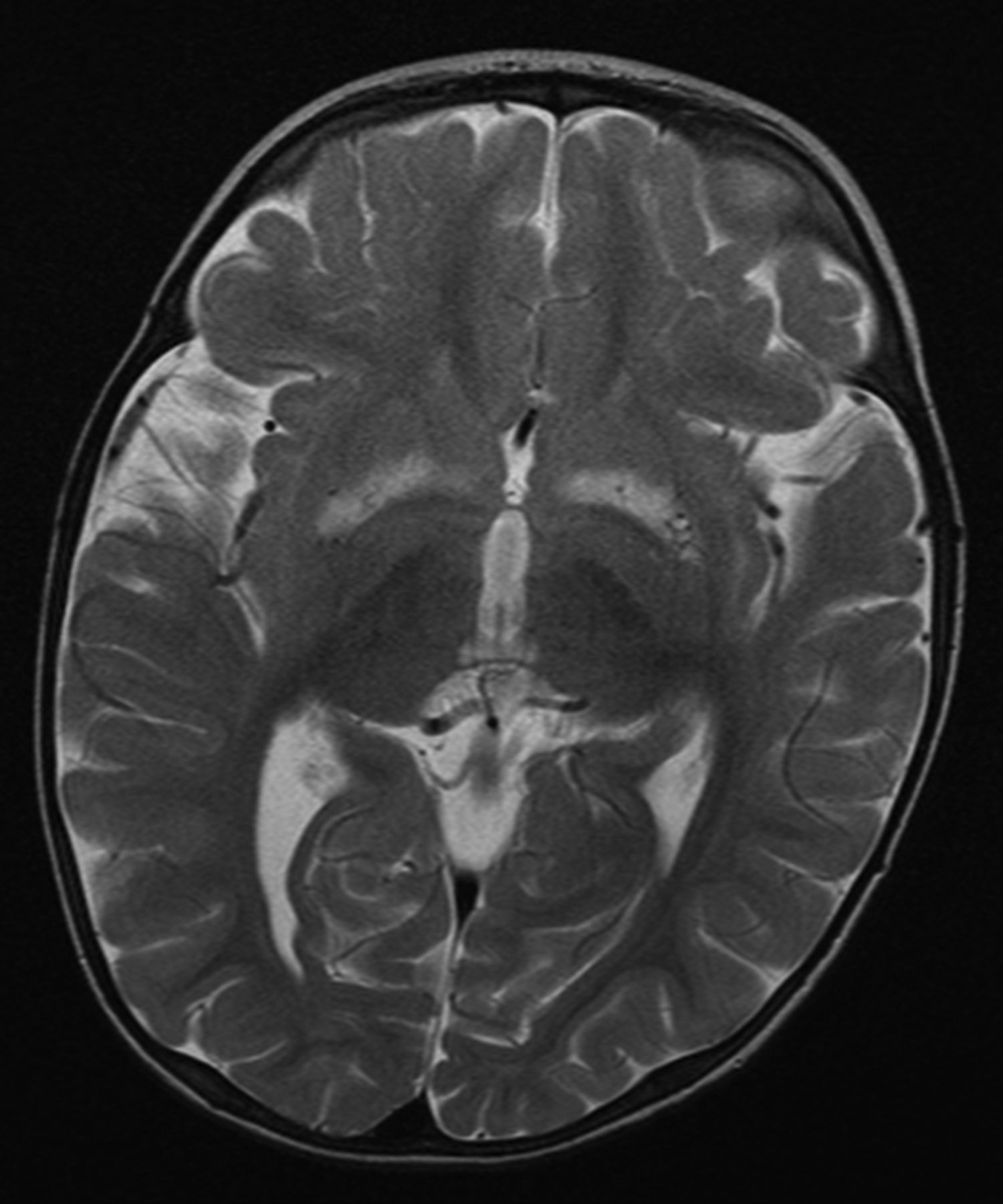
Bilateral, symmetric basal ganglia lesions on MRI in a patient with Leigh's syndrome due to the m.9176T>C mutation in mitochondrial DNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mitochondria in human skin fibroblasts. In these cells the mitochondria form a number of networks. Each network is colour-coded: the short networks are yellow and longer networks are red. (A) and (B) show the mitochondrial networks from two separate healthy controls. (C) and (D) show the longer networks seen in fibroblasts from two different patients with compound heterozygous mutations in SPG7, which tend to be longer than those found in healthy controls. (Adapted from Pfeffer et al.31)
When compared with idiopathic Parkinson's disease, patients with mitochondrial disease who develop parkinsonism typically have additional clinical features. In Huntington's disease and in several very rare neuroacanthocytosis syndromes, the chorea is commonly associated with other symptoms related to neurodegeneration, and so they may resemble mitochondrial disease.55 ,56 In Wilson's disease, a copper metabolism disorder caused by mutations in the ATP7B gene, parkinsonism and dystonia are among the clinical features.57 Segawa's disease, a dopa-responsive dystonia caused by mutation of GCH1, is characterised by a progressive dystonia and a sustained, marked response to treatment with levodopa.58 It is important that clinicians should promptly recognise both Wilson's disease and Segawa's disease, as there is useful medical treatment for both conditions. In contrast to these two disorders, movement disorders in the context of mitochondrial disease usually have other features that suggest conditions such as optic atrophy, myoclonus, neuropathy or cerebellar ataxia.
Gastrointestinal abnormalities
Gastrointestinal system abnormalities are increasingly recognised as mitochondrial disease manifestations.59 Mitochondrial neurogastrointestinal encephalomyopathy syndrome (MNGIE) is an autosomal recessive condition caused by mutations in the thymidine phosphorylase gene TP. MNGIE is clinically relatively homogeneous, and typical gastrointestinal symptoms include recurrent diarrhoea, abdominal pain and cramps, early satiety and intestinal pseudo-obstruction.60 Neurological features of MNGIE include peripheral neuropathy, PEO and hearing loss. Cerebral imaging typically shows leukodystrophy, and mtDNA studies identify multiple mtDNA deletions or mtDNA depletion. Reduced activity of thymidine phosphorylase in leukocytes is diagnostic. Several homozygous or heterozygous TP mutations have been reported in patients with MNGIE.60
Gastrointestinal symptoms in the form of bloating, dysphagia, anorexia, chronic diarrhoea and pseudo-obstruction are also prevalent among patients with the m.3243A>G mutation, and are associated with profound COX deficiency and high levels of mutation heteroplasmy in gastrointestinal smooth muscle.59 In a Japanese study, 40% of patients with the m.3243A>G mutation were affected by chronic intestinal pseudo-obstruction,61 which was sometimes the presenting clinical feature.
Patients with MNGIE are usually exceptionally thin.60 The additional features of PEO, hearing loss and leukodystrophy in brain imaging comprise a syndromic phenotype that suggests a mitochondrial disease. In the case of the m.3243A>G mutation, common clinical features suggesting a mitochondrial disorder are diabetes mellitus, hearing loss, PEO, and in some cases, stroke-like episodes.62 Isolated gastrointestinal symptoms are uncommon in patients with mitochondrial disease.
Mitochondrial disease ‘chameleon’ presentations: clinical considerations
Low specificity of several common features
Several clinical features that are prevalent in mitochondrial disease, such as diabetes mellitus, epilepsy, migraine-like headache, peripheral neuropathy or sensorineural hearing loss, have low specificity regarding the diagnosis of a mitochondrial disorder. This presents a clinical challenge because these features may be the presenting feature of a mitochondrial disease, but look very similar to more ‘common or garden’ disorders. However, isolated features of exercise intolerance, gastrointestinal dysmotility, myopathy and parkinsonism are uncommon in mitochondrial disease, although they may be encountered as part of a syndromic phenotype.
Oligosymptomatic patients
Patients with mitochondrial disease do not necessarily present with a typical syndromic phenotype with several affected organ systems, but can be oligosymptomatic. The full picture may only emerge over time, or after a detailed family history is taken.
Missing or misleading inheritance pattern
Although mitochondrial disease is typically inherited either maternally or in an autosomal dominant or recessive pattern, it can also arise de novo in a family. Moreover, mtDNA point mutations are often heteroplasmic, meaning that only some of a patient's mtDNA are mutated. As heteroplasmy levels may vary between individuals and generations in the maternal line, it is possible that an inherited mtDNA mutation does not cause any symptoms in the close maternal relatives of a patient. As a consequence, many patients with mitochondrial disease do not have a relevant family history. This is also often the case for autosomal recessive mitochondrial disorders in an outbred population.
Combination of the above
Consider a 35-year-old man with recently diagnosed, insulin-dependent diabetes mellitus who reports no relevant family history. The patient's diabetes might be due to him harbouring the m.3243A>G mutation, but because of the oligosymptomatic phenotype, a common presenting phenotype, and the ‘missing’ maternal family history suggesting a mitochondrial disease, reaching the right diagnosis can be very challenging. Under these circumstances, further investigation of a mitochondrial disorder is mandatory.
Conclusions
In mitochondrial disease there are large overlaps between the clinical manifestations of various genetic defects, a wide variety of phenotypes are caused by the same genetic defect, and many individual features are also common in the general population (eg, diabetes mellitus, migraine). This means that the possibility of a mitochondrial disorder might be entertained in a huge number patients every day in the general neurology clinic. Generally speaking, the combination of multiple pathologies affecting several organ systems, especially those relying greatly on intact oxidative energy production, should lead the neurologist to consider the possibility of an underlying mitochondrial disorder (box 3). The all-important starting point is to raise the suspicion of a mitochondrial disease at the right moment. This triggers appropriate clinical investigations that will define the full phenotype, and lead on to appropriate biochemical and molecular genetic investigations to provide a definitive diagnosis.
When to suspect mitochondrial disease—some suggestions
Clinical features compatible with well-established mitochondrial syndromes such as CPEO, MELAS, Leber's hereditary optic neuropathy, MERRF or NARP
PEO, irrespective of inheritance pattern
Combination of diabetes mellitus and sensorineural hearing loss, with the onset of both typically in young adult age (∼18–45 years)
Syndromic, multisystem disorder with any combination of features prevalent in mitochondrial disease (eg, ataxia, diabetes mellitus, epilepsy, myopathy, neuropathy, pigmentary retinopathy, sensorineural hearing loss); sometimes but not always with a maternal inheritance pattern
Severe (predominantly occipital) epilepsy with onset in adolescence or young adult age, often accompanied by migraine-like headaches, ataxia or cerebral white matter changes in brain MRI (especially in the cerebellum and thalamus), and epilepsia partialis continua—POLG-related epilepsy. Caveat: avoid sodium valproate-induced liver failure.
Myopathy with histopathological evidence of COX-negative fibres and/or ragged-red fibres exceeding the number expected with normal ageing (ie, there should be no ragged-red fibres/COX-negative fibres present in <50-year olds, and <5% in those aged over 50 years)
COX, cytochrome c oxidase; CPEO, chronic progressive external ophthalmoplegia; MELAS, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; MERRF, myoclonic epilepsy with ragged-red fibres; NARP, neuropathy, ataxia and retinitis pigmentosa; PEO, progressive external ophthalmoplegia.
The traditional approach to diagnosing mitochondrial disorders in adults has relied on identifying characteristic clinical syndromes, and gathering evidence from histological and histochemical studies of a muscle sample, and from biochemical analysis of the respiratory chain complex activities. These preliminary investigations have been complemented by targeted molecular genetic studies to detect mutations in mtDNA or nuclear DNA genes, secondary mtDNA rearrangements or mtDNA depletion.63 However, this rather laborious process is now being challenged by ‘next generation’ sequencing approaches that are already being implemented in clinical practice.64 These technological advances will make the molecular diagnostics of mitochondrial disorders much more straightforward than previously.
Key points
Mitochondrial disease may present with a multitude of clinical features in different combinations; the most energy-dependent organs of the body such as the brain, heart, skeletal muscles and endocrine pancreas, are commonly affected.
Most patients with a mitochondrial disease do not show the ‘full house’ of features typical of the canonical mitochondrial clinical syndromes.
Although common neurological disorders, such as migraine, exercise intolerance, parkinsonism, epilepsy, ataxia and dementia, can occur in patients with mitochondrial disease, these are rare in isolation.
Many patients with mitochondrial disease do not have a relevant family history; this is particularly often the case for autosomal recessive mitochondrial disorders in an outbred population.
‘Next generation’ sequencing approaches are already being implemented in clinical practice and will make the molecular diagnostics of mitochondrial disorders much more straightforward than previously.
References
Footnotes
Contributors MHM and PFC wrote the article. MHM carried out the literature search and wrote the first draft. Both authors revised the article and are responsible for the final version.
Funding MHM is funded by the Sigrid Jusélius Foundation. PFC is an Honorary Consultant Neurologist at Newcastle upon Tyne Foundation Hospitals NHS Trust, a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z) and a UK NIHR Senior Investigator. PFC receives additional support from the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease research (G0601943), and EU FP7 TIRCON and the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed. Reviewed by Nick Wood, London, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Diagnosis and therapy in neuromuscular disorders: diagnosis and new treatments in mitochondrial diseases
- SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions
- Recognition, investigation and management of mitochondrial disease
- Could it be mitochondrial? When and how to investigate
- Clinical mitochondrial genetics
- Human extraocular muscles in mitochondrial diseases: comparing chronic progressive external ophthalmoplegia with Leber’s hereditary optic neuropathy
- Risk of cardiac manifestations in adult mitochondrial disease caused by nuclear genetic defects
- Novel POLG1 mutations associated with neuromuscular and liver phenotypes in adults and children
- Republished: A practical approach to late-onset cerebellar ataxia: putting the disorder with lack of order into order
- Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) in the older adult