Article Text

Abstract
Rituximab is a widely used B-cell-depleting monoclonal antibody. It is unlicensed for use in neurological disorders and there are no treatment guidelines. However, as a rapidly acting, targeted therapy with growing evidence of efficacy and tolerability in several neuroinflammatory disorders, it is an attractive alternative to conventional immunomodulatory medications. This practical review aims to explain the basic principles of B-cell depletion with therapeutic monoclonal antibodies. We present the evidence for using rituximab in neurological diseases, and describe the practical aspects of prescribing, including dosing, monitoring, safety, treatment failure and its use in special circumstances such as coexisting viral hepatitis, pregnancy and lactation. We provide an administration guide, checklist and patient information leaflet, which can be adapted for local use. Finally, we review the safety data of rituximab and ocrelizumab (a newer and recently licensed B-cell-depleting therapy for multiple sclerosis) and suggest monitoring and risk reduction strategies.
- rituximab
- ocrelizumab
- Bcell
- monoclonal antibody
Statistics from Altmetric.com
Introduction
This article covers both the practical aspects of prescribing rituximab and some of the basic principles of B-cell depletion with monoclonal antibodies, which are relevant to neurologists. Those seeking an administration guide for rituximab, or a rapid overview of the indications and supporting evidence, expected side-effects or specific prescribing circumstances, should skip to the relevant tables towards the end of the article. We have provided an example of a patient information sheet and an administration checklist, which are available as online supplementary material 1 and 2.
Approach to managing symptomatic secondary antibody deficiency
This advice is appropriate for patients satisfying all three of the following criteria:
Maintenance rituximab therapy.
Serious or recurrent (particularly respiratory) infections.
Total serum IgG <6.0 g/L (recurrent infection is more likely if IgG <4.0 g/L).
Suggested management to mitigate infection risk:89
Liaise with local immunology service.
Check disease-specific circulating antibody titres against Haemophilus influenzae (Hib), Clostridium tetani and pneumococcal capsular polysaccharide.
If titres are below protective cut-off levels (Hib >1 mcg/mL, tetanus >0.1 IU/mL, pneumococcus >50 mg/L), 90 vaccinate patients and retest titres after 6 weeks.
Trial prophylactic antibiotic therapy.
Immunoglobulin replacement therapy (IGRT) is justifiable if the response to test vaccination and/or antibiotics is poor.91
– Initiate intravenous immunoglobulin at 0.4–0.6 g/kg/month or consider subcutaneous formulations.
– Aim for serum IgG within normal range (6–16 g/L).
Assess clinical response to IGRT after 6 months (burden of infections) and consider the need for long-term treatment. IGRT is unlikely to reduce the frequency of urinary tract infections.
B-cell function and role in neurological disease
B-cells secrete antibodies, present antigen and regulate the immune response by producing proinflammatory and anti-inflammatory cytokines. Only 2.5% of the total B-cell population is within the peripheral circulation, made up predominantly of naïve mature B-cells and memory B-cells; the rest are in bone marrow and lymphoid tissue.1 Antibodies may be of any immunoglobulin class (G, M, A, D or E) or subclass (eg, IgG1–4), each of which have differing functions. Examples of disorders in which autoantibodies are almost certainly pathogenic include myasthenia gravis with acetylcholine receptor (AChR) antibodies (usually IgG1 or IgG3) or muscle-specific tyrosine kinase (MuSK) antibodies (IgG4), neuromyelitis optica spectrum disorders (NMOSD) with antibodies against the aquaporin-4 water channel (mainly IgG1), and autoimmune encephalitis with antibodies to the N-methyl-D-aspartate receptor (NMDAR) (mainly IgG1) or leucine-rich glioma inactivated-1 (LGI1) (mainly IgG4). B-cells also play a crucial role in multiple sclerosis (MS) pathogenesis, evidenced by cerebrospinal fluid oligoclonal IgG bands, meningeal-based ectopic B-cell follicles adjacent to areas of focal cortical demyelination2 and the efficacy of B-cell-depleting therapies to treat MS.
B-cell surface markers
CD19 and CD20 are B-cell transmembrane proteins. They can be used as targets for drugs and as surface markers (in flow cytometry to quantify B-cell populations and assess treatment response). CD19 is expressed more widely throughout B-cell development than CD20 but both markers are absent on long-lived plasma cells (figure 1). In healthy adults CD19+ or CD20+ B-cells comprise 12%–22% of the total circulating lymphocyte population (absolute reference range is 50–500 cells/mm3).
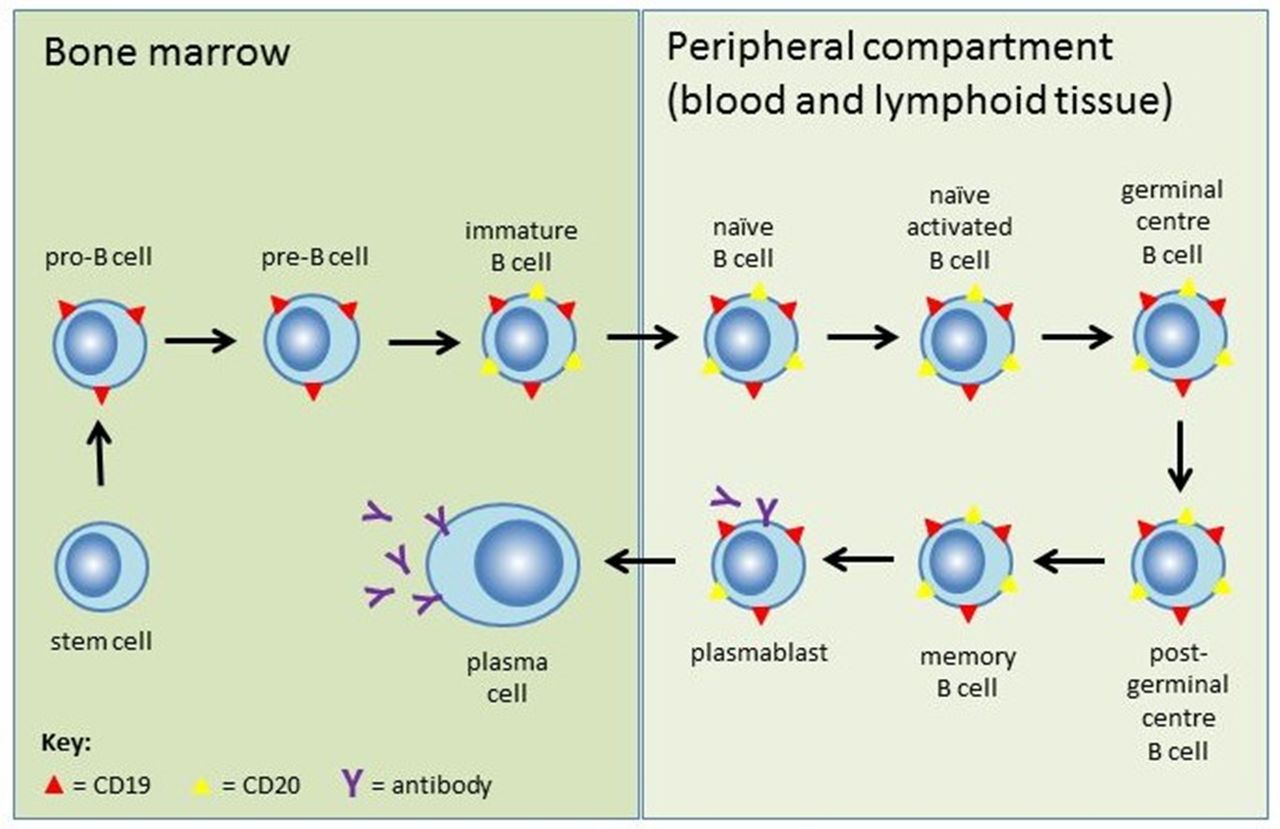
Stages of B-cell development and expression of B-cell surface markers. Pluripotent haematopoietic stem cells develop into naïve mature B cells in the bone marrow. They then migrate to secondary lymphoid organs (spleen and lymph nodes), where they are activated by antigens in circulating lymph and mature into memory B-cells or plasmablasts. Memory B-cells either circulate in the bloodstream or remain in germinal centres, while plasmablasts mature to antibody-secreting plasma cells that reside in the bone marrow or lymphoid tissue. CD20 (yellow triangles) appears at the immature B-cell stage and is lost at the plasmablast stage. Most plasmablasts and nearly all plasma cells (which produce the vast majority of antibodies) do not express CD20. CD19 (red triangles) has wider expression from the pro-B-cell stage through to plasmablasts and a proportion of plasma cells, but not terminally differentiated plasma cells.
CD27 is expressed by memory B-cells and certain other immune cell types. The combination of CD19 and CD27 is specific to memory B-cells. This subset of long-lived B-cells, capable of rapid differentiation into high-affinity plasma cells following repeated antigen exposure, may be an important target in the treatment of autoimmune neurological disease.3 4
B-cell-depleting monoclonal antibodies
Monoclonal antibodies are immunoglobulins produced by a single clone of hybridoma cells (antigen-specific plasma cells fused with myeloma cells). They bind via their two identical fragment antigen binding (Fab) domains to a single epitope and activate the immune system via their fragment crystallisable (Fc) domain. Cells expressing that epitope are killed, therefore allowing highly targeted immunotherapy for a variety of neoplastic and autoimmune diseases. Available B-cell-depleting monoclonal antibodies have Fab domains targeted to CD20 or CD19, and so selectively deplete the circulating B-cell population, with the exception of mature antibody-secreting plasma cells. figure 2 shows those used in treating neuroinflammatory diseases.

B-cell-depleting monoclonal antibodies in neurology. mAb, monoclonal antibody; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorder.
Rituximab was the first anti-CD20 monoclonal antibody to be approved (1997) for treating B-cell lymphomas. It has since been licensed to treat refractory rheumatoid arthritis and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Unlicensed use for neuroinflammatory disease is growing.
Rituximab is a first-generation, chimeric monoclonal antibody made by fusing a murine (rodent) Fab domain with a human Fc domain (‘chimeric’ is from the mythological Chimera—a monstrous fire-breathing hybrid creature, part lion and part goat). The Fc domain activates various immune mechanisms, as shown in figure 3. Ninety per cent of circulating B-cells are killed within 3 days of the first infusion of rituximab. Reduction of pathogenic antibody titres correlates with efficacy in some disorders. However, rituximab probably affects the whole spectrum of B-cell function, and secondary changes in T-cell function, such as induction of immunoregulatory T cells, may be important in some neuroinflammatory disorders. Sparing of CD20negative long-lived plasma cells is hoped to preserve lasting humoral immunity.

Rituximab depletes CD20+ B-cells via three different mechanisms: (1) antibody-dependent cellular cytotoxicity mediated by Fcγ receptors on the surface of natural killer cells, granulocytes and macrophages; (2) complement-dependent cytotoxicity; (3) induction of apoptosis.
Compared with first-generation monoclonal antibodies, second-generation monoclonal antibodies have improved Fab domains, often humanised or fully human, which improve B-cell killing and tolerability (figure 2). Ocrelizumab (humanised) was recently approved to treat relapsing and progressive MS. Ofatumumab, a fully human monoclonal antibody given by once monthly subcutaneous injection, is in clinical trials. Third-generation monoclonal antibodies have been further engineered to improve their Fc-mediated immune functions or half-life. Ublituximab (TG-1101), a rapidly infusible chimeric glycoengineered monoclonal antibody, is also being trialled in MS currently.
Anti-CD19 B-cell-depleting therapies may be more effective (and potentially have higher risks) than anti-CD20 therapies due to the broader expression of CD19 throughout B-cell development, including the plasmablast phase (figure 1). Inebilizumab (MEDI-551) is in a phase 3 trial in NMOSD.5
Biosimilars
Most monoclonal antibodies are costly. However, once the original drug patent expires, cheaper, copy versions—‘biosimilars’—become available. Competing companies do not have access to the original molecular clone, cell bank or exact manufacturing process, which may result in slight differences to these complex molecular structures. Therefore, biosimilars are not truly ‘generic’. To gain a licence, biosimilars must be shown to be highly similar in structure, purity and biological activity to the original monoclonal antibody; however, it is not necessary to repeat clinical trials for each indication. Rituximab’s patent expired in 2016 and the European Medicines Agency (EMA) has approved two biosimilars, Truxima and Rixathon. The dosing and administration protocols are identical. British National Formulary prices are currently £1746 for MabThera 1 g (the original form of rituximab) vs £1572 for Truxima or Rixathon.6 However, prices to National Health Service (NHS) hospitals vary substantially according to regional contracts and discussion with the hospital pharmacy department is advised. Patients should be informed of the switch and monitored to ensure that tolerability and side effects remain unchanged.
Indications and evidence for rituximab in neurology
An understanding of the evidence for rituximab in neuroinflammatory disorders (see table 1 for a briefer summary) should inform off-license prescribing.
Indications for rituximab in neurology
Multiple sclerosis
With a choice of licensed disease-modifying therapies supported by phase III randomised controlled trials, use of rituximab in the UK for MS is rare. However, there is evidence suggesting efficacy, and it may be an option in occasional cases (especially if licensed comorbidities, such as active rheumatoid arthritis, facilitate funding). Phase I and II trials of rituximab in relapsing–remitting MS met their primary endpoints.7–9 A large 96-week multicentre randomised controlled trial in primary progressive MS failed to demonstrate a delay to confirmed disease progression, but subgroup analysis showed a benefit in younger patients, particularly with inflammatory lesions.10 Trials in MS then ceased, probably due to the impending expiration of rituximab’s patent and the emergence of newer B-cell-depleting therapies from the same manufacturer. Sweden is the biggest off-licence prescriber of rituximab for all forms of MS and has published class IV evidence of safety and efficacy in a large multicentre cohort (n=822).11 The dose used is 500–1000 mg 6–12 monthly. A recent real-world retrospective comparative study showed efficacy in relapsing–remitting MS comparable to natalizumab and fingolimod, and significantly better than injectable disease-modifying therapies and dimethyl fumarate. Rituximab was superior to all drugs in terms of discontinuation rate.12 Although this is relatively low-quality evidence, there is a clear indication that rituximab is an effective treatment for MS, which would be expected in light of the recent positive randomised controlled trials for ocrelizumab.
Neuromyelitis optica spectrum disorders
No immunosuppressive therapy in NMOSD is yet validated by a high-quality randomised controlled trial, though there are three such trials ongoing. Rituximab use is supported by numerous, predominantly retrospective, case series amounting to over 400 patients and showing consistent reductions in annualised relapse rate. There are various dosing strategies in use, which we discuss later in ‘dosing and monitoring’. A recent meta-analysis calculated a mean reduction in relapse rate of 79%.13 As such, rituximab currently has the best evidence of any immunotherapy used in NMOSD, but due to its relatively high cost, it remains second-line therapy for patients in the UK. It is available for patients who have relapsed despite adequate treatment with azathioprine or mycophenolate mofetil combined with low-dose prednisolone.14 Funding can be obtained through the Specialised NHS England Service for NMOSD (www.nmouk.nhs.uk).
Autoimmune encephalitis
As most autoimmune encephalitis is monophasic, the role of rituximab is usually as a second-line acute therapy (single course) to maximise neurological recovery, rather than as a long-term maintenance treatment (as with MS/NMOSD). The most commonly used dosing regimen is 375 mg/m2 weekly for four doses. Limited retrospective evidence supports its use when there has been an inadequate response to intravenous corticosteroids, plasma exchange and intravenous immunoglobulin. There is no evidence to compare the effects of individual immunotherapies in autoimmune encephalitis, so it is not possible to ascribe therapeutic benefits solely to rituximab. However, its rapid onset of action, established efficacy in other antibody-mediated diseases and good safety profile with short-term use make it an attractive option. The major study supporting rituximab use in autoimmune encephalitis is a retrospective comparison of outcomes in 161 patients. Functional improvement measured by modified Rankin Scale occurred more frequently in the rituximab-treated group, regardless of antibody status).15
There is additional evidence specifically for anti-NMDAR encephalitis, the most common subtype of autoimmune encephalitis. A large prospective cohort study (n=577) found that 78% of patients who failed first-line and received second-line immunotherapy (rituximab and/or cyclophosphamide) had a good outcome at 24 months, compared with 55% of patients who failed first-line and did not receive second-line therapy.16 A study of rituximab in paediatric neuroinflammatory disease included 44 patients with anti-NMDAR encephalitis. Ninety-seven per cent of these patients had some benefit from second-line rituximab therapy, especially when given early.17 In light of these studies, a UK clinical commissioning policy, published in March 2018, agreed to fund rituximab routinely for adults and children with anti-NMDAR encephalitis who have responded inadequately to first-line therapy (failure to improve by two or more points on the modified Rankin Scale by 4 weeks from starting first-line treatment or by 6 weeks from symptom onset).18
Evidence for autoimmune encephalitis with less common antibodies is limited to case reports and small case series, which are frequently confounded by coadministration of multiple immunotherapies. For example, there are two case series reporting outcomes after rituximab in seven patients with anti-LGI1 encephalitis. Three patients (43%) had good outcomes and one patient had a possible response.19 20 The emerging theme in autoimmune encephalitis, irrespective of antibody status, is that early and aggressive immunotherapy is beneficial. It seems plausible that rituximab, or similar B-cell-depleting therapies, will increasingly form part of immunotherapy algorithms.
Primary angiitis of the central nervous system
High-dose corticosteroids with or without cyclophosphamide form the mainstay of treatment for this rare condition.21 Favourable outcomes with rituximab are reported in two small case series, in which 2/2 and 6/7 patients appeared to respond.22 23 There are additional case reports describing its use.24
ANCA-associated vasculitis
ANCA-associated vasculitis occasionally presents to the neurologist, for example, with mononeuritis multiplex, but is likely to be comanaged with other vasculitis experts. Rituximab is licensed and recommended by recent European Guidelines for organ or life-threatening disease.25 This follows two randomised controlled trials, in which rituximab (375 mg/m2 weekly for four doses) was non-inferior to cyclophosphamide for inducing remission.26 27 It may be more effective than cyclophosphamide for relapsing disease.27 NHS England will fund rituximab where cyclophosphamide has failed or is contraindicated (eg, patients who wish to preserve their reproductive potential).28
Stiff-person syndrome
Although some case reports suggested a possible benefit of rituximab for stiff-person syndrome,29–31 a single small double-blind randomised controlled trial (n=24) found no significant changes in any outcome measures after 6 months of rituximab treatment.32
Immune-mediated peripheral neuropathies
A UK clinical commissioning policy, published in December 2017, reviewed the evidence for rituximab to treat chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), multifocal motor neuropathy, non-systemic vasculitic neuropathy and IgM paraprotein-associated demyelinating neuropathy with antibodies to myelin-associated glycoprotein (anti-MAG neuropathy). It concluded that there is insufficient evidence to make rituximab routinely available for these disorders.33 However, there may be circumstances in which rituximab could help, as discussed below. Most studies have used 375 mg/m2 weekly for 4 weeks.
Rituximab has been used in CIDP following inadequate response to conventional therapy (corticosteroids, intravenous immunoglobulin and plasma exchange). A Cochrane review (2013) identified 17 published CIDP cases treated with rituximab, of which 12 (71%) improved after treatment.34 The largest series has 10 patients, of whom six (60%) improved.35 In a multicentre retrospective analysis, 18/110 (16.4%) refractory CIDP cases received rituximab. The response rate (improvement in modified Rankin Scale score by at least 1 point) was 33%—comparable to azathioprine or cyclophosphamide.36 There was a recent report of marked improvement following rituximab in patients with CIDP with IgG4 antibodies against paranodal proteins (anti-neurofascin 155/CNTN1). These cases account for less than 10% of all patients with CIDP but they are often relatively resistant to intravenous immunoglobulin and corticosteroids, highlighting the importance of serological testing and suggesting a potential role for rituximab in a subset of patients with CIDP that needs further exploration.37 38
Data for rituximab in multifocal motor neuropathy are limited to small case series and are conflicting. Intravenous immunoglobulin is the mainstay of therapy. When rituximab was used as monotherapy in seven patients in two separate observational studies, all showed some improvement in muscle strength.39 40 When given as an adjunct to intravenous immunoglobulin in a small open-label trial (n=6), there was no significant change in motor function or required dose.41 In two further cases, one patient reduced and one increased their intravenous immunoglobulin requirement.42
Non-systemic vasculitic neuropathy is a peripheral nerve vasculitis in the absence of clinical or laboratory evidence of systemic vasculitis. The Peripheral Nerve Society guideline (2010) lists rituximab as an unproven treatment option, favouring high-dose corticosteroids and escalation to cyclophosphamide if needed.43 Rituximab could possibly be considered on an individual funding basis in patients with refractory non-systemic vasculitic neuropathy, on the basis of its efficacy in ANCA-associated vasculitis.44
Two placebo-controlled trials of rituximab for anti-MAG neuropathy showed marginal benefits. In the first study, 4/13 (31%) rituximab-treated patients improved by one or more Inflammatory Neuropathy Course and Treatment (INCAT) score compared with 0/13 placebo-treated patients (p=0.036).45 In the second study (n=54), there was no significant difference in the absolute INCAT sensory score between the groups (negative primary outcome), but the number of patients with improvement in INCAT disability score was higher in the rituximab-treated group.46 Several prospective observational studies report improvements in roughly half to two-thirds of patients.47–50
Myasthenia gravis
International consensus guidelines (2016) advise that ‘rituximab should be considered as an early therapeutic option in patients with MuSK-associated myasthenia gravis who have an unsatisfactory response to initial immunotherapy.’51 A formal consensus could not be reached for AChR-associated myasthenia gravis. Several predominantly retrospective, observational studies and two systematic reviews have investigated rituximab as an acute therapy (usually a single course with variable dosing) for refractory myasthenia gravis (persistent weakness or need for high-dose corticosteroids despite conventional immunosuppression).
Despite many case series being shared between the systematic reviews, the reported response rates in AChR-associated myasthenia gravis are discordant, with 30%–80% of patients achieving a Myasthenia Gravis Foundation of America post-intervention status (MGFA-PIS) of ‘minimal manifestations or better’ following rituximab.52 53 This may be explained by variability in patient selection, inclusion of many ‘burnt out’, unresponsive cases and inclusion of cases where MGFA-PIS was not used as an outcome measure in the original report. Response did not correlate well with AChR antibody titres.53 Two ongoing randomised controlled trials may help better define the efficacy of rituximab in AChR-associated myasthenia gravis in the near future.
In comparison, response rates in MuSK-associated myasthenia gravis were high (72%–89%) in both reviews.52 53 A further blinded prospective review found 67% of rituximab-treated patients obtained MGFA-PIS of ‘minimal manifestations or better’ versus 26% of controls.54 The benefit of rituximab in MuSK-associated myasthenia gravis appears to be more prolonged and correlates better with antibody titres.53 55 MuSK antibodies are of the IgG4 subtype whereas AChR antibodies are of the IgG1/3 subtype. The superior efficacy of rituximab may therefore be explained by selective depletion of short-lived IgG4-producing B-cells.55
Dosing and monitoring of rituximab
Rituximab is given by intravenous infusion over 3–6 hours. A solution for subcutaneous injection is available but is not used in neurology and therefore will not be discussed in this review. There is no validated dosing strategy for rituximab in neuroinflammatory disease and there is great heterogeneity in the literature. Figure 4 is a suggested administration guide. The two most common dosing regimens are either 375 mg/m2 body surface area given once weekly for 4 weeks (adopted from haemato-oncology) or two infusions of 500–1000 mg given a fortnight apart (adopted from clinical trials in rheumatoid arthritis). Following two 1000 mg infusions, the mean half-life of rituximab is 20.8 days (range 8.58–35.9 days).56

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rituximab administration guide. Italicisedpoints reflect our personal practice rather than established recommendations. HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; NMDAR, N-methyl-D-aspartate receptor; NMOSD, neuromyelitis optica spectrum disorders; TB, tuberculosis; VZV, varicella zoster virus.
In rheumatoid arthritis there is no significant difference in the clinical responses after high-dose (2×1000 mg) and lower dose (2×500 mg) rituximab regimens.57 The clinical response correlates with the degree of B-cell depletion, not the rituximab dose used.58 The same is likely to be true in neuroinflammatory disease. Doses as low as 100 mg weekly for 3–4 weeks have been used successfully in small series of patients with MS, NMOSD and anti-NMDAR encephalitis.59–62
Near complete B-cell depletion occurs within a fortnight of infusion and usually persists for 6–12 months. Therefore, where maintenance treatment is planned, repeated courses have commonly been given at regular six monthly intervals. However, patients vary significantly in both the initial rituximab dose required to achieve B-cell depletion and the time to B-cell repopulation. In a study of patients with NMOSD, 17% repopulated their B-cells before 6 months.63 Prolonged B-cell depletion lasting over 3 years following a single dose of rituximab is also reported.64 This makes a case for monitoring and retreating according to B-cell repopulation, which will identify ‘early repopulators’ at risk of disease relapse, and limit overtreatment of patients with sustained B-cell depletion, thereby preventing complications and reducing cost.
Although rituximab is an anti-CD20 antibody, quantification of CD19+ cells using flow cytometry is the preferred method for monitoring B-cell depletion and repopulation. This is because rituximab still present in serum could block binding of fluorophore-labelled anti-CD20 antibodies used in flow cytometry, thereby interfering with the detection of B-cells.
Among the several relapsing illnesses that may benefit from rituximab, relapses from NMOSD pose the highest risk of permanent disability. However, the critical threshold of B-cells in the measurable peripheral circulation that is associated with NMOSD relapse is undetermined and is likely to vary with the disease and individual. Neurologists have retreated when the CD19+ B-cell count becomes detectable65 or more than 0.1% of total circulating lymphocyte count.66 Some measure the much smaller memory B-cell (CD19+/CD27+) population (see figure 4 —option 2).4 67 Switching from six monthly infusions to memory B-cell-monitored treatment reduces cumulative rituximab dose without apparent loss of efficacy.68 However, standardisation of flow cytometry techniques and inaccuracy when quantifying very small cell populations can pose problems.69 In the UK NMO Service we use monthly CD19+ B-cell monitoring and have found 1% (an arbitrary value based on clinician experience) to be an acceptable cut-off for retreatment for the majority of patients.70 In those who relapse with a detectable B-cell count below 1%, retreatment aiming for complete suppression is suggested before considering treatment failure and switching immunotherapy.
Treatment failure
Where treatment failure is suspected, we advise excluding alternative possibilities, such as intercurrent infection, and ensuring that B-cell depletion is adequate by checking a peripheral blood CD19+ B-cell count. Possible reasons for treatment failure include the following:
Lack of efficacy of B-cell depletion
In a large NMOSD cohort (n=100), nine patients (9%) experienced relapses despite CD19+/CD27+ memory B-cell depletion within target range.71 NMOSD relapses occurring on rituximab are generally milder than those occurring off treatment. Non-circulating B-cells in lymphoid tissues (ie, most of the total body B-cell population) and long-lived plasma cells are not thought to be depleted by rituximab and may have a role in breakthrough disease.
Early relapses/delayed therapeutic onset
Early NMOSD relapses can follow rituximab induction therapy.4 72 73 This may be due to incomplete B-cell depletion. Alternatively, initial B-cell depletion may induce release of systemic B-cell activating factor, promoting autoantibody production by plasma cells, and ‘leading to a transient rise’ in antibody titre and early relapses.74
Incomplete B-cell depletion/early repopulators
Genetic factors may explain why some patients do not maintain adequate B-cell depletion. These include polymorphisms in the B-cell activating factor gene or in the Fc gamma receptor 3A gene expressed by the effector cells that mediate B-cell killing (figure 3).71 75 Another hypothetical reason might be the development of antidrug antibodies.
Antidrug antibodies
The efficacy of some monoclonal antibodies is reduced by antidrug antibodies, for example, anti-tumour necrosis factor agents. Fab binding could have a neutralising effect and Fc binding may increase drug clearance. However, the role of anti-drug antibodies in rituximab treatment failure is uncertain. They were identified in a third of patients with MS treated with rituximab.76 They may have a greater effect in patients on low-dose rituximab (100 mg infusions)77 but higher, standard doses probably overcome the effects of antidrug antibodies.76 78 Outside of trials, detection of antidrug antibodies can be technically difficult, poorly standardised and is hard to obtain for routine use.
Combination with other immunosuppressive medications
Due to the risk of early relapse after rituximab initiation, some neurologists continue moderate-dose prednisolone (usually 10–20 mg daily) for 4–12 weeks in NMOSD. The decision to continue corticosteroids depends on the condition being treated and individual patient factors.
Combination with other immunosuppressive medications can be considered in some circumstances but must be balanced against the risk of immunocompromise. We generally reserve combination therapy for refractory disease. In treating rheumatoid arthritis, rituximab is often combined with methotrexate or leflunomide but there is little evidence to guide practice in neuroinflammatory disease.
Risks and adverse events
The efficacy of rituximab and current safety data support its use, and the longer term safety profile will become clearer with increasing use of B-cell-depleting therapies like ocrelizumab. Tables 2 and 3 summarise the approach to adverse events and special prescribing circumstances. Italicised points denote personal practice, rather than established recommendations.
Other considerations and special circumstances when prescribing rituximab
Rituximab treatment risks and management. Unless a separate reference is given, information is adapted from MabThera SmPC [56], experience from RA
The relatively favourable safety profile of rituximab is likely due to preservation of protective antibody production by CD20negative long-lived plasma cells. However, it remains uncertain whether long-term humoral immunity results entirely from these self-sustaining cells or whether replenishment of plasma cells by memory B-cells is required. Several studies have reported secondary antibody deficiency complicating rituximab therapy—a risk that appears to increase with repeated courses and lower pretreatment levels of immunoglobulins.67 78–80 Not all patients with hypogammaglobulinaemia develop infections, but we recently reported a series of serious sinopulmonary infections associated with hypogammaglobulinaemia occurring in patients with NMOSD on long-term rituximab.81 All patients had prior exposure to immunosuppressant medications. This has led to changes in our practice, with greater focus on pretreatment vaccinations, B-cell monitoring to limit cumulative rituximab dose and targeted use of immunoglobulin replacement therapy to mitigate sinopulmonary infections in selected patients (see figure 4, table 3 and Box 1).
Pregnancy and breast feeding
Rituximab crosses the placenta after 20 weeks’ gestation. Although not known for certain, the existing evidence suggests that rituximab is possibly safe for use during early pregnancy (see table 2).82 The prolonged B-cell-depleting effect (sometimes greater than the 40 weeks of gestation) can be used advantageously. For example, in planned pregnancies, rituximab could be given before conception and after delivery, sparing the gestating fetus from B-cell depletion.
In relapsing conditions with high morbidity, such as NMOSD, the risk of relapse during protracted interruption of rituximab therapy for conception and pregnancy is a dilemma for many women. A recent expert review suggests that two doses of 1000 mg could be given as close as 1 month before planned conception in the hope that B-cell depletion will persist for the duration of pregnancy. They advise that rituximab could be resumed in the first week after delivery given the very high postpartum risk of NMOSD relapse.83 However, women should be counselled about the limited data on rituximab-exposed pregnancies.84
Ocrelizumab
While this is review is primarily intended to cover rituximab, it may be remiss not to discuss ocrelizumab, as this is the first anti-CD20 therapy to gain a licence (Food and Drug Administration, EMA) for a neurological indication (MS). Ocrelizumab has been in development for over a decade but progress in rheumatoid arthritis was halted in 2010 after data from multiple phase III trials suggested an excess of serious infections and a poor benefit-risk profile when combined with methotrexate. However, trials in MS continued and it was licensed in the USA in March 2017 and in Europe in January 2018. The European licence is for treating active relapsing MS and early primary progressive MS with imaging features of inflammatory activity. Recent phase III randomised controlled trials showed that ocrelizumab reduced annualised relapse rates versus interferon beta-1a in relapsing MS, and reduced 12-week confirmed disability progression versus placebo in primary progressive MS.85 86 The trials used a fixed dosing schedule over 2 years of follow-up. The safety profile appeared favourable. Infusion reactions were frequent but rarely problematic. Upper respiratory tract infections were more common after ocrelizumab but there was no excess of serious or opportunistic infections. Ocrelizumab was associated with low total serum IgM in 16% of patients, but no increased infection risk was observed in these patients. There was no reduction in total serum IgG or disease-specific antibody titres over the 2-year follow-up period. An increased risk of malignancies (including breast cancer) was observed in the ocrelizumab trial arms but the incidence was within the background rate expected for an MS population.85 86
Ocrelizumab has been licensed as a fixed six-monthly dosing regimen with no specific immune function monitoring, despite the fact that considerable interindividual variation is observed in time-to-repopulation of B-cells following ocrelizumab.87 The experience of ocrelizumab in clinical trials may seem inconsistent with our and others’ real-world experience of rituximab, in which we have observed the coexistence of secondary antibody deficiency and increased rate of infections in patients with NMOSD on maintenance therapy.78–81 We postulate that this may relate to a degree of baseline immune dysfunction caused by prior immunosuppressive medication and a longer treatment duration than is recorded in the pivotal ocrelizumab studies. This echoes experience in vasculitis, where previous immunosuppressive therapy (particularly cyclophosphamide) has been identified as a risk factor for greater decline in immunoglobulin levels and more prolonged B-cell depletion after rituximab.25 88 In contrast, the vast majority of patients recruited to the MS ocrelizumab trials were treatment naïve or had used non-immunosuppressive disease-modified therapies. Safety information on ocrelizumab from postmarketing surveillance will be useful to further inform risk and to guide whether flexible dosing may become preferable in certain situations. Sequential treatment effects following high-efficacy disease-modified therapies are also yet to be explored.
Conclusion
Rituximab is a valuable treatment option for a variety of neuroinflammatory conditions. While there are no randomised controlled trials and questions remain about optimal dosing strategies, there is a growing body of evidence to support its use in specific situations. Overall, rituximab has an excellent safety profile, and relative to other immunomodulatory treatments, it may be an option for managing severe active diseases in pregnancy. However, neurologists need to be aware of specific management issues, including secondary antibody deficiency in patients requiring maintenance B-cell depletion. Specific risk factors to consider include low pretreatment immunoglobulin levels, prior use of immunosuppressive drugs or a requirement for ongoing combination therapy.
Newer and more costly B-cell-depleting therapies show additional promise in recent and ongoing trials but it remains to be seen if more effective and prolonged B-cell depletion will pose additional risks. Prospective registries with extended follow-up will be important in better defining the real-life risks and benefits for patients.
Supplemental material
Supplemental material
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.
- 93.
- 94.
- 95.
- 96.
- 97.
- 98.
- 99.
- 100.
- 101.
- 102.
- 103.
- 104.
- 105.
- 106.
Footnotes
Contributors DHW drafted the manuscript and subsequent revisions. All authors critically appraised, revised the manuscript for important intellectual content and approved the final version of the article. AJ conceived and designed the review.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests ECT has received honoraria and support to attend educational meetings from Merck, support to attend educational meetings from Biogen and salary as a UK MS Registry fellow from Biogen. SJ has received advisory board, consulting, meeting attendance, speaker, study, author and project support from CSL Behring, Shire, LFB, Biotest, Binding Site, UCB Pharma, Grifols, Octapharma, SOBI, GSK, Sanofi, BPL, Zarodex, Weatherden and Uptodate. SH has previously received funding from the NIHR Oxford Biomedical Research Centre, the Watney Trust and Myaware. RJM has acted as consultant to, or received support for speaking at or chairing meetings, or received grant funding for research from AKL Pharma, BMS, Cellgene, Chugai, Eli Lilly, Novartis, Pfizer, Roche, Sandoz, Sanofi and UCB Pharma. HJK has received speaking and/or consulting support from Bayer Schering Pharma, Biogen, Celltrion, Eisai, HanAll BioPharma, MedImmune, Merck Serono, Novartis, Sanofi Genzyme, Teva-Handok and UCB; research support from Ministry of Science & ICT, Sanofi Genzyme, Teva-Handok and UCB. He is a steering committee member for MedImmune, and coeditor/associated editor of MS Journal-Experimental, Translational and Clinical, and Journal of Clinical Neurology. NPR has received personal fees and other from Biogen, grants from Novartis, grants and other from Genzyme, Roche and Teva, and personal fees from Merck. BACC has received personal compensation for consulting for AbbVie, Biogen, EMD Serono, GeNeuro, Novartis and Sanofi Genzyme. AJ has received compensation for advisory board, consulting, meeting attendance and speaking from Biogen, Terumo-BCT, Genentech, Shire and Chugai Pharmaceuticals.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed by Jackie Palace, Oxford, UK; Neil Scolding, Bristol, UK; and Jon Sussman, Manchester, UK.
Other content recommended for you
- Anti-CD20 therapies in pregnancy and breast feeding: a review and ABN guidelines
- Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders
- Neurodegeneration in multiple sclerosis and neuromyelitis optica
- Autologous haematopoietic stem cell transplantation for neurological diseases
- Anti-CD20 therapies decrease humoral immune response to SARS-CoV-2 in patients with multiple sclerosis or neuromyelitis optica spectrum disorders
- Autologous haematopoietic stem cell transplantation for immune-mediated neurological diseases: what, how, who and why?
- A practical guide to the treatment of neuromyelitis optica
- Neuromyelitis optica spectrum disorder presenting as rhomboencephalitis
- Humoral and cellular responses to SARS-CoV-2 vaccination in patients with autoantibody-mediated neuroimmunology
- Clinical relevance of distinguishing autoimmune nodopathies from CIDP: longitudinal assessment in a large cohort