Article Text

Statistics from Altmetric.com
A 38-year-old woman gave a 1-year history of difficulties climbing stairs and walking long distances, needing a wheelchair at times. More recently, she had also experienced weakness of her arms with difficulties doing her hair or lifting light weights. She reported shortness of breath on mild exertion and had lost 12 pounds in weight because of muscle loss. She had no muscle pain, episodes of dark urine, muscle twitching, visual disturbances, hearing loss, speech difficulties, dysphagia, numbness or tingling, bladder or bowel difficulties, fevers, rash or joint pain. She had met all milestones at a normal age and had normal development.
Her medical history was unremarkable. She had not taken any lipid-lowering agents and there was no illicit drug use or toxic exposure. There was no relevant family history and no consanguinity.
On examination, she showed bilateral and symmetric scapular winging. There was no percussion or action myotonia or paramyotonia, and no fasciculations. Her muscle bulk and tone were normal. Cranial nerves were all normal. Manual muscle strength testing showed the following Medical Research Council (MRC) grades (right/left where applicable): neck flexion 4–, neck extension 5, shoulder abduction 4/4, external rotation of elbow 4/4, elbow flexion 4/4, elbow extension 4+/4+, wrist flexion 5/5, wrist extension 5/5, finger flexion 5/5, finger extension 5/5, hip flexion 4/4, hip extension 3/3, hip abduction 3/3, knee extension 4+/4+, knee flexion 4/4, ankle dorsiflexion 5/5 and ankle plantar flexion 5/5. Deep tendon reflexes were 1+ at triceps, brachioradialis and ankles, and 2+ at biceps and knees bilaterally. She had a waddling gait. Plantar responses were flexor. Sensory and coordination exams were normal.
Question 1: what is the differential diagnosis?
This 38-year-old woman likely has a myopathy given the symmetrical pattern of proximal weakness involving legs and arms. The differential diagnosis includes an acquired (e.g., inflammatory or toxic myopathy) or genetic (e.g., muscular dystrophy, late-onset congenital myopathy or metabolic myopathy) myopathic disorder. A neuromuscular junction disorder needs to be considered although the absence of ocular symptoms (which occurs in 90% of patients with myasthenia) and lack of fluctuating symptoms would be atypical for myasthenia gravis, and the lack of autonomic symptoms and intact patella reflexes would be atypical for Lambert-Eaton myasthenic syndrome. A motor neurone disease such as spinal muscular atrophy type III is unlikely since muscle weakness typically progresses more slowly than in this case.
Question 2: what investigations would you like to perform?
Serum creatine kinase concentrations were persistently elevated, ranging from 682 to 1464 U/L (reference range, 26–192). Full blood count, serum electrolytes, anti-nuclear antibody, anti-neutrophil cytoplasmic antibodies, thyroid-stimulating hormone, erythrocyte sedimentation rate, C reactive protein, serum protein electrophoresis with immunofixation, hepatitis B and C serology, and myositis-specific antibody panel were all normal or negative. She had already undergone electromyography and a muscle biopsy of right gastrocnemius muscle before our initial evaluation. That electromyography had shown abnormal spontaneous activity in the form of positive sharp waves and fibrillation potentials in right thoracic paraspinal and right medial gastrocnemius muscles, without any apparent abnormality in motor unit morphology or recruitment. The muscle biopsy of right gastrocnemius muscle was normal. Also, a transthoracic echocardiogram was normal.
Question 3: how would you interpret these test results?
Fibrillation potentials and positive sharp waves are not specific since they can develop in either a neuropathic or a myopathic process with muscle membrane irritability (inflammatory, toxic/necrotic, dystrophic, and selected congenital and metabolic myopathies). Morphology of motor unit potentials (high amplitude and long duration in neurogenic disorders and low amplitude and short duration in myopathies) and recruitment (reduced in neurogenic and early in myopathic disorders) would help to distinguish a myopathic from a neuropathic process, but there were none of these abnormalities on this electromyography study. The right gastrocnemius muscle had full strength on examination, which significantly reduces the diagnostic yield of a biopsy; an optimal muscle to biopsy should be clinically weak but not end stage. Thus, we continue to favour a myopathy in her case based on history, examination and high serum creatine kinase. The most likely diagnosis is an adult-onset limb-girdle muscular dystrophy, given scapular winging—which is not typical of acquired myopathies—and her pattern of weakness. Less commonly, some congenital myopathies (e.g., central core) can present like this in adulthood. Finally, metabolic myopathies need to be considered. She had no exercise intolerance with myoglobinuria to suggest a dynamic form of glycogen storage disease such as McArdle's disease. However, others such as an adult-onset Pompe’s disease (glycogenosis type II) may manifest with progressive proximal weakness, together with mild-to-moderately elevated serum creatine kinase, and myopathic features on electromyography involving thoracic paraspinal muscles. It is important to consider Pompe’s disease as there is now treatment in the form of enzyme replacement therapy. Finally, lipid storage myopathies, which are rarer, can present in a similar fashion. Whereas a dynamic form of lipid storage myopathy (such as mitochondrial trifunctional protein deficiency, very-long-chain acyl-CoA dehydrogenase deficiency and carnitine palmitoyl transferase II deficiency) is unlikely, static and progressive forms within this group need to be considered.
Question 4: how would you proceed now?
We tested for serum acid α-glucosidase activity to screen for Pompe’s disease and performed genetic testing for the most common forms of limb-girdle muscular dystrophy (ANO5, CAPN3, CAV3, DMD, DES, DNAJB6, DYSF, FKRP, FKTN, GAA, GMPPB, LMNA, MYOT, POMGNT1, POMT1, POMT2, SGCA, SGCB, SGCD, SGCG, TCAP, TNPO3, TRIM32 and TTN), which were all normal. We then performed an electromyography that showed increased insertional activity and abnormal spontaneous activity in the form of positive sharp waves and fibrillation potentials in left deltoid and biceps muscles, with early recruitment of short-duration, low-amplitude and polyphasic motor unit potentials. On the same day, we performed a muscle biopsy of right deltoid to avoid potential needle insertion–related inflammatory changes present on the left (figure 1).
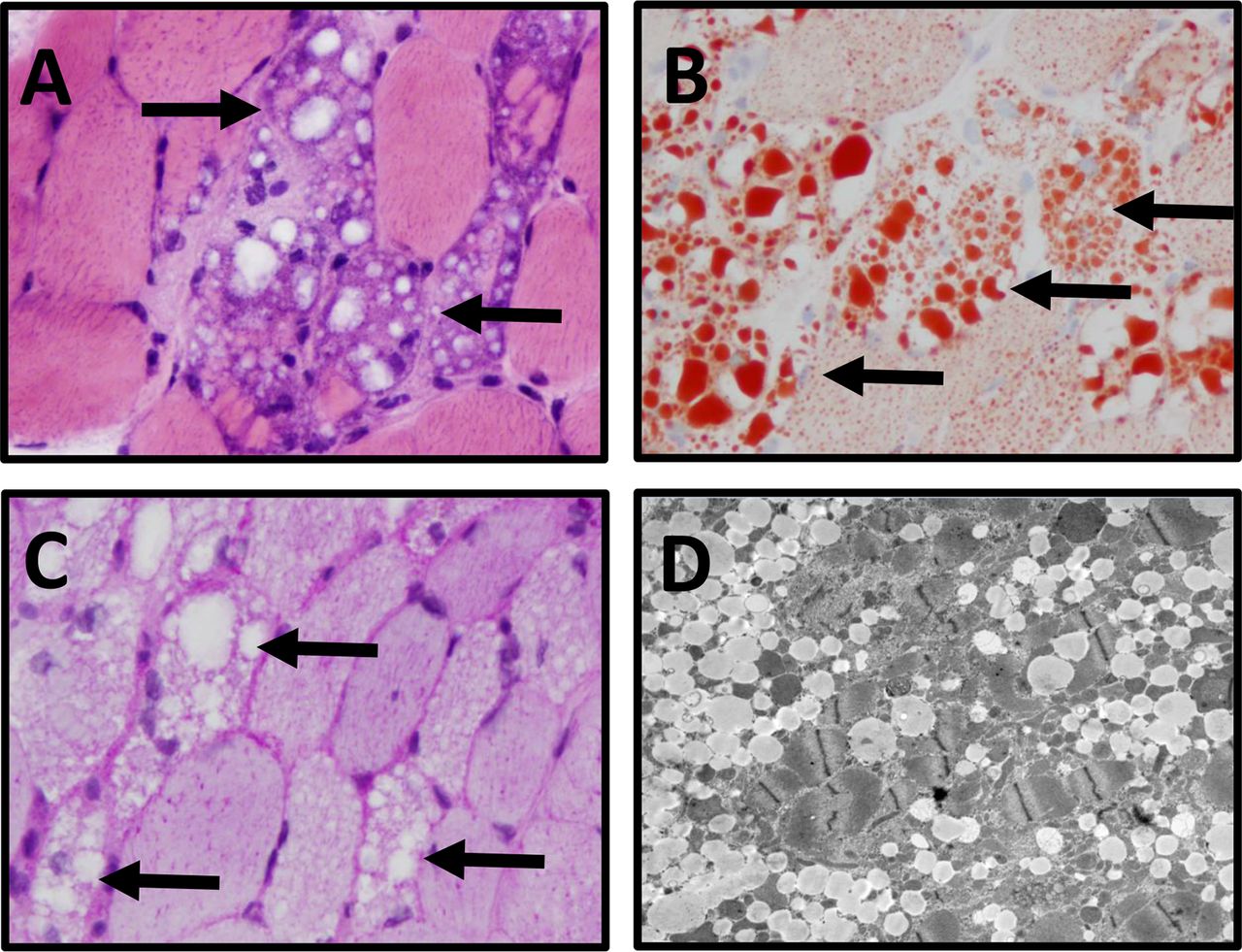
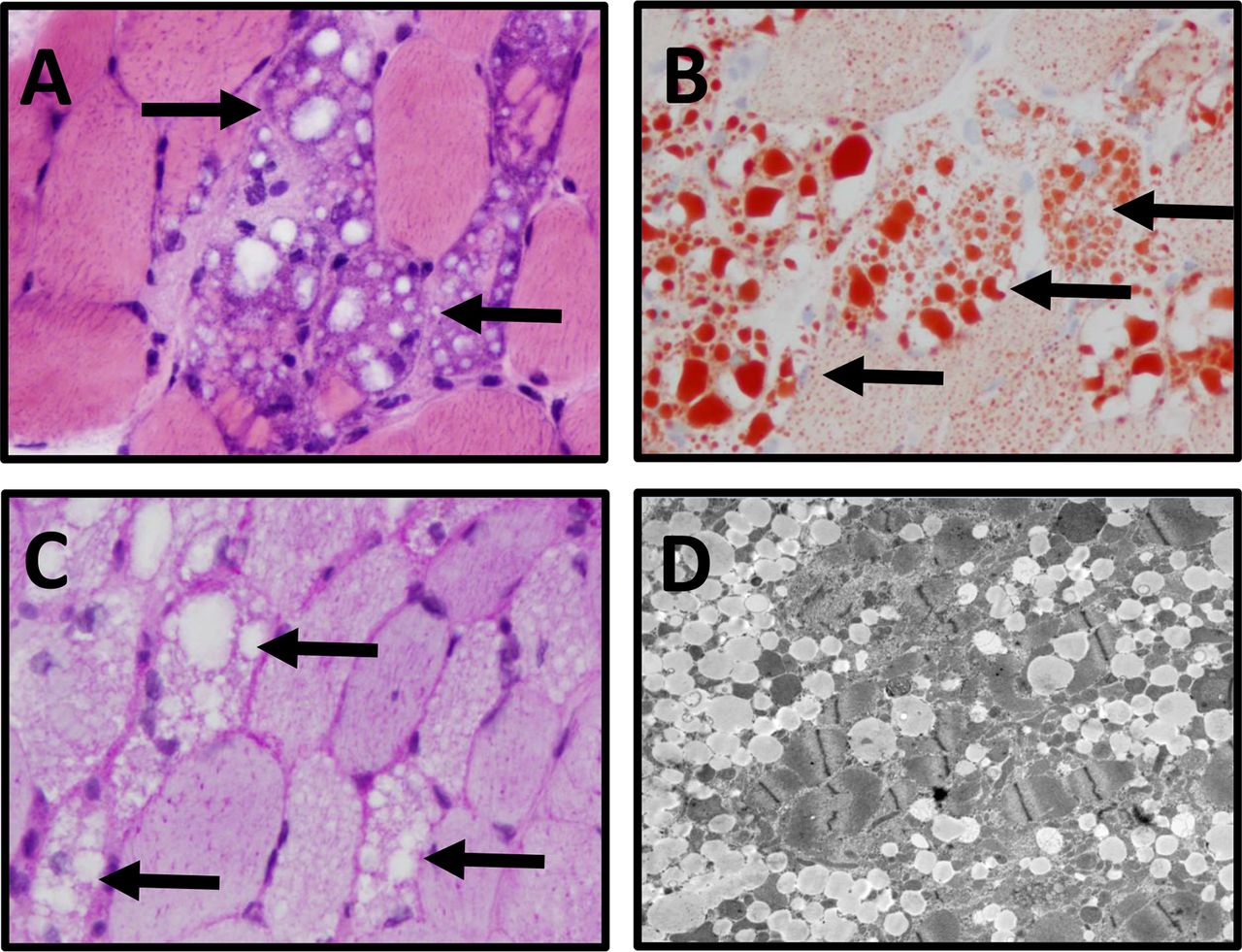
Muscle biopsy of the right deltoid. The right deltoid muscle biopsy shows (A) numerous vacuolated myofibres (black arrows) (frozen section H&E, ×400); (B) the cytoplasmic vacuoles contain lipid (arrows) (oil red O stain, ×400) but (C) not glycogen (arrows) (PAS stain, 400x); (D) electron microscopy images show myofibres with abundant intracellular myocyte lipid vacuoles.
Question 5: which are the most useful investigations to reach final diagnosis?
This patient has a lipid-storage myopathy with massive accumulation of lipids in muscle fibres, which can develop in primary carnitine deficiency, multiple acyl-CoA dehydrogenase deficiency (MADD) or neutral lipid storage disease. These diseases commonly manifest as progressive proximal muscle weakness rather than as episodic exercise intolerance. Free and total carnitine concentrations in serum, acylcarnitines in serum (its diagnostic yield increases following a 12-hour fasting period) and organic acids in urine will help to distinguish between these three conditions. In primary carnitine deficiency, serum free and total carnitine, and serum acylcarnitines would be markedly reduced whereas urine organic acids would be normal. In MADD, short (C4–C8), medium (C8–C12) and long (>C12) acylcarnitines would be all elevated in serum, free and total carnitine can be normal or secondarily reduced, and organic acids would be increased in urine. In neutral lipid storage disease, serum free and total carnitine and acylcarnitines, and urine organic acids would be all normal because the accumulated lipids are triglycerides and not fatty acids.
Her serum-free carnitine concentration was mildly reduced at 19 µmol/L (normal: 25–60) and total carnitine concentration was 23 µmol/L (normal: 5–29), which ruled out a primary carnitine deficiency. However, the acylcarnitine profile showed high concentrations of short, medium and long acylcarnitines, and organic acids in urine showed elevation of 2-hydroxyglutaric acid. This biochemical profile is characteristic of MADD.
Question 6: how would you confirm diagnosis and treat this patient?
MADD is inherited in an autosomal recessive manner and most patients (93%) harbour mutations in ETFDH; this gene encodes an electron transfer flavoprotein–ubiquinone oxidoreductase (ETF-QO) in charge of transferring beta-oxidation-derived electrons to respiratory chain in mitochondria. Also, most patients respond to riboflavin supplementation.
Genetic testing of ETFDH identified two variants in this patient: c.814G>A, p.Gly272Arg and c.1204A>G, p.Thr402Ala. Her strength and function dramatically improved after 3 weeks of taking riboflavin (400 mg/day). Thus, she did not need her wheelchair anymore, she did not feel winded while walking and she could stand up from a chair without using her arms. At 3-month follow-up, her examination was improved in the following muscle groups (MRC grades, right/left where applicable): neck flexion 4+, shoulder abduction 5/5, elbow external rotation 5/5, elbow flexion 5/5, elbow extension 5/5, hip extension 4/4, hip abduction 4/4, knee extension 5/5, and knee flexion 5/5. Her serum creatine kinase normalised to 89 U/L (reference range, 26–192) and serum concentrations of acylcarnitines either normalised or decreased at 3 months post-treatment.
We advised her to continue taking riboflavin 400 mg/day, which we suspect she will need for life.
Commentary
MADD (also called glutaric aciduria type II) is an autosomal recessive disease caused by mutations in ETFDH (93%) and ETFA/B (~7%) that halt the transfer of electrons from the beta-oxidation of short-chain, medium-chain, and long-chain fatty acids to the respiratory chain in mitochondria. As a result, all different length-chain fatty acids are increased in serum in the form of acylcarnitines, and organic acids in urine are elevated, pointing to a failure of fatty acid oxidation.1
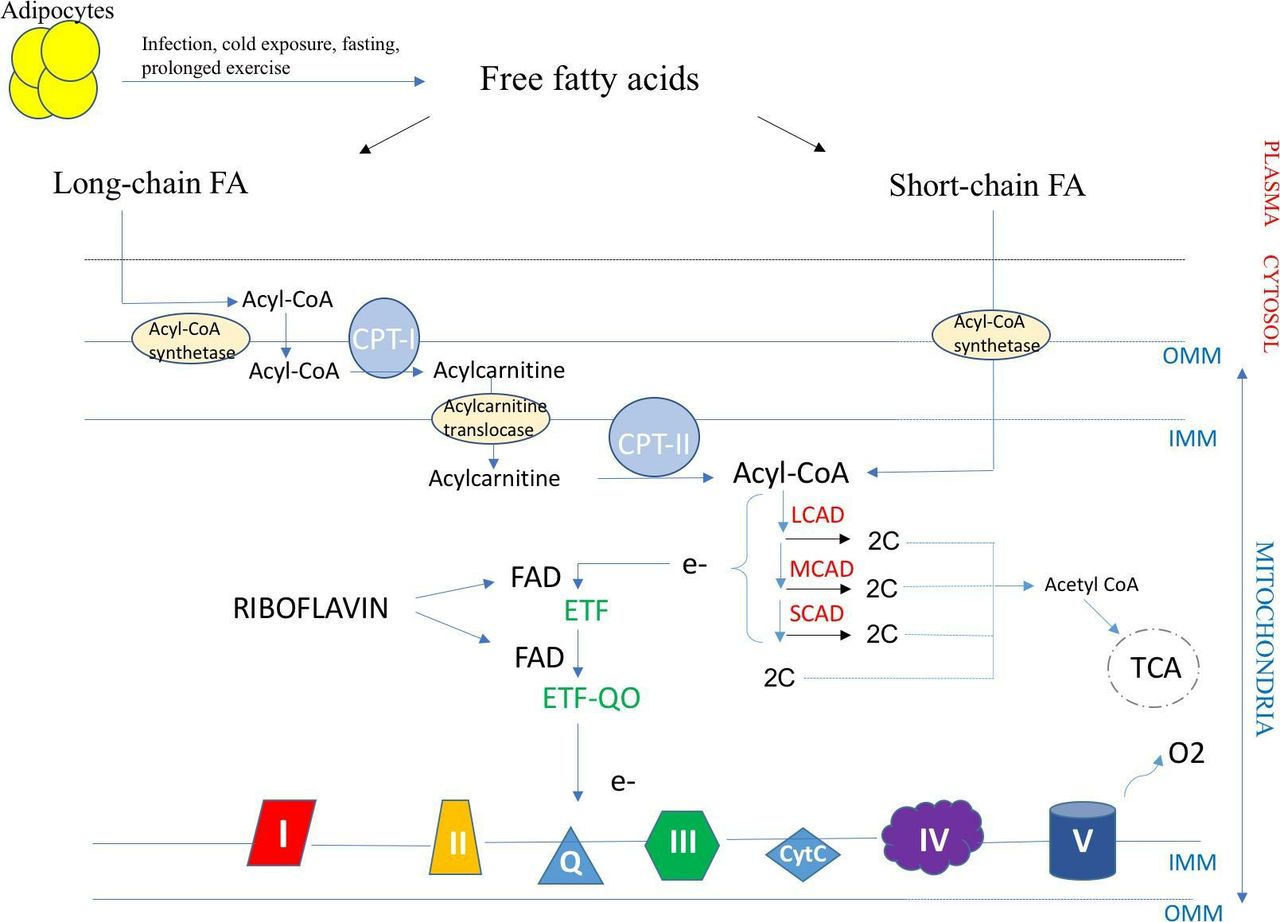
Riboflavin (or vitamin B2) is a precursor of flavin adenine dinucleotide, which is a required cofactor of the flavoproteins encoded by E ETFDH (ETF-QO) and ETFA/B (ETF) (figure 2). Riboflavin supplementation at a dose of 100–400 mg/day has been reported in small series to be effective in MADD.1
{kind=link}
{kind=link}
Fatty acid transport and β-oxidation in the mitochondria. The transport of long-chain fatty acids into the mitochondria is CPT complex dependent unlike short-chain fatty acids. LCAD, MCAD and SCAD are enzymes in charge of the β-oxidation of acyl-CoA which releases electrons that are transferred to ETF (encoded by ETFA and ETFB), then ETF-QO (encoded by ETFDH), and lastly to mitochondrial respiratory chain. FAD is a cofactor of both ETF and ETF-QO. Riboflavin is a precursor of FAD and the mainstay treatment of MADD due to ETFDH and ETFA/B mutations. CPT I and II, carnitine palmitoyl transferase I and II; ETF, electron transfer flavoprotein; ETF-QO, electron transfer flavoprotein–ubiquinone oxidoreductase; FA, fatty acid; FAD, flavin adenine dinucleotide; IMM, inner mitochondrial membrane; LCAD, long-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA; OMM, outer mitochondrial membrane; SCAD, short-chain acyl-CoA dehydrogenase; TCA, tricarboxylic acid cycle.
The MADD phenotype varies greatly in different age groups; the milder phenotype occurs in adulthood in the form of proximal myopathy, as in the patient here presented. The neonatal form is often fatal leading to multiorgan failure. Although rare in Western countries, it is probably underdiagnosed. A normal serum creatine kinase, electromyography study, serum acylcarnitine profile or urine organic acids (which may occur under metabolically stable conditions) do not rule out MADD. Although not specific, the MR scan may show fat infiltration and muscle atrophy in the posterior compartment of thighs and gluteal muscles.2 However, diagnosis of MADD mainly relies on muscle biopsy, which shows massive accumulation of lipid droplets within muscle fibres and confirmatory genetic testing. We would like this report to raise awareness of this myopathy, which in most cases responds spectacularly to riboflavin supplementation, and also to review the differential diagnosis of lipid-storage myopathies (table 1).3 4
Differential diagnosis of lipid-storage myopathies
Footnotes
Contributors PG-P drafted the initial manuscript, revised the final manuscript and was involved in the clinical care of the patient. MT and JH revised the final manuscript, performed pathological studies and provided figure. AAA drafted the manuscript, revised the manuscript until final version, and was involved and supervised the clinical care of the patient.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests PG-P reports no disclosures. MT reports no disclosures.JH reports no disclosures. AAmato served on Scientific Advisory Boards or was a medical consultant for Novartis, Acceleron, Akashi, Idera and Alexion. He is also a medical consultant for Best US Doctors. He receives publishing royalties for his book Neuromuscular Disease from McGraw-Hill. He was a co-investigator on clinical trials sponsored by Novartis, Idera, Acceleron and Alexion. He receives an honorarium from the American Academy of Neurology as Associate Editor of Neurology.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned. Externally peer reviewed by Jon Walters, Swansea, UK.
Linked Articles
- Editors’ commentary
Other content recommended for you
- Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations
- Metabolic myopathies: a practical approach
- Sensory neuropathy as a manifestation of multiple acyl-coenzyme A dehydrogenase deficiency
- Newborn screening for primary carnitine deficiency: who will benefit? – a retrospective cohort study
- Tandem mass spectrometry findings at autopsy for detection of metabolic disease in infant deaths: postmortem changes and confounding factors
- Progressive gait disturbance with head drop
- A diagnostic approach to recurrent myalgia and rhabdomyolysis in children
- Screening for medium chain acyl-CoA dehydrogenase deficiency using electrospray ionisation tandem mass spectrometry
- Do renal and cardiac malformations in the fetus signal carnitine palmitoyltransferase II deficiency? A rare lethal fatty acid oxidation defect
- 2823 MADD muscles: a case report of very-late onset multiple acyl-CoA dehydrogenase deficiency (MADD)