Article Text

Abstract
Sarcoidosis affects the nervous system in 10% of cases. When it does so it can affect any part of the nervous system and with all degrees of severity. It forms part of the differential diagnosis in inflammatory, infective, neoplastic and degenerative neurological diseases and may be very difficult to diagnose without histological confirmation. Recent clinical studies and the increasing availability of new biological treatments allow a much clearer understanding of the disease. This review summarises its clinical features, imaging and laboratory characteristics, treatment and outcome.
- neuroimmunology
Statistics from Altmetric.com
Introduction
Sarcoidosis is an auto-inflammatory disorder characterised by the development of granulomatous inflammation in affected tissues. Any tissue may become affected, the most frequently being the lungs, skin, eyes and liver. The lymphatic system is always involved. At onset 95% of cases have demonstrable lung involvement, and half have multisystem disease1; over time this prevalence increases in those whose disease does not spontaneously remit.2
Clinical features
The onset is subacute, rarely acute, with constitutional symptoms including feeling unwell, fever and night sweats before the heralding symptoms arise. Lofgren’s syndrome is the coexistence of fever, arthritis of the ankles, erythema nodosum and hilar lymphadenopathy (figure 1). Heerfordt’s syndrome comprises a granulomatous uveitis, parotid and submandibular salivary gland swelling and cranial neuropathy (usually but not always the facial nerve). Lung involvement is characterised by cough, breathlessness and chest tightness or stabbing pains, but is often asymptomatic. Any structure within the eye may be involved, and skin, cardiac, hepatic, renal, bone and joint involvement is common.3

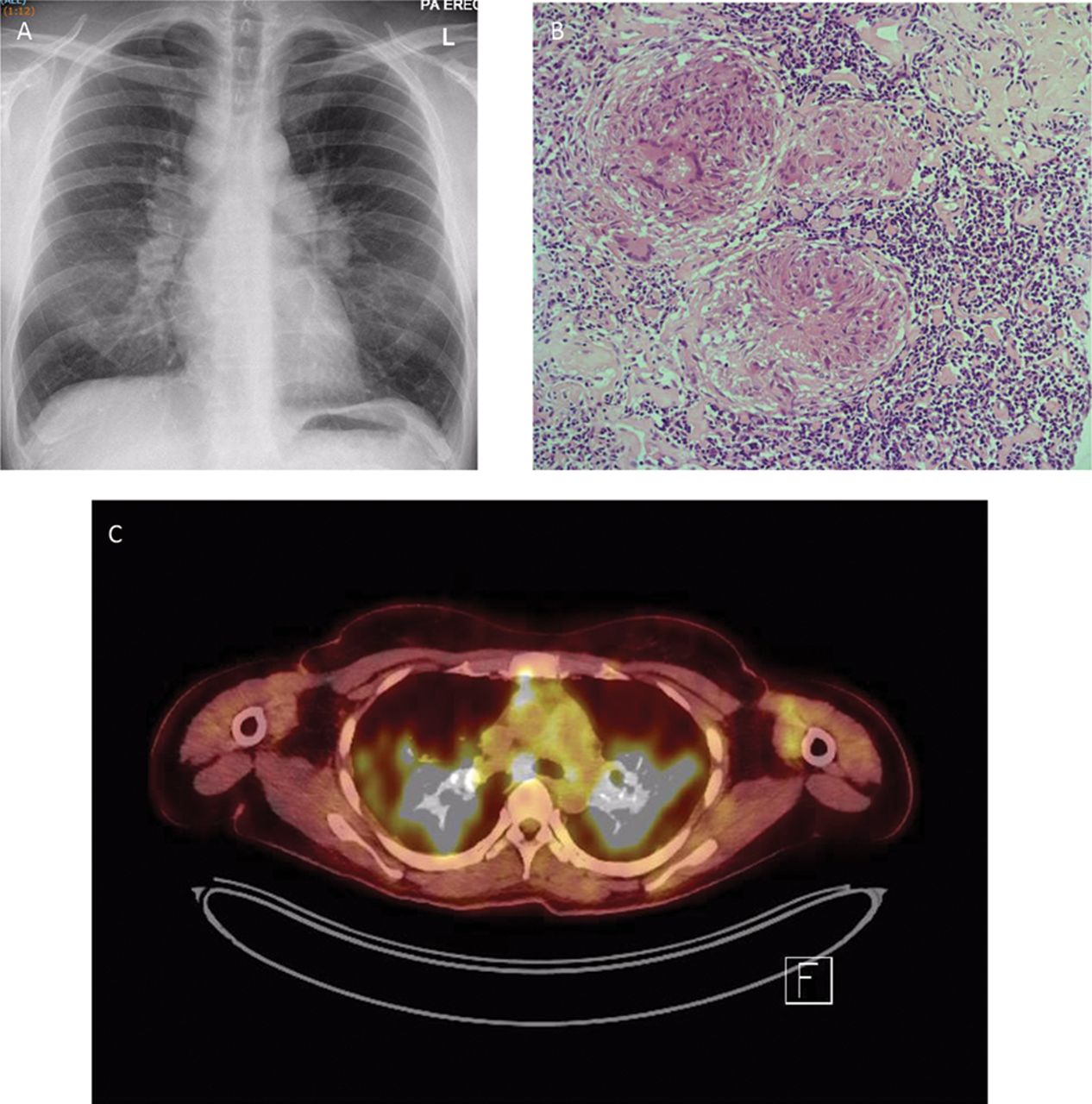
(A) Chest X-ray showing Garland’s triad of bilateral symmetrical hilar lymphadenopathy and right para-tracheal lymphadenopathy. (B) H&E-stained section of mediastinal lymph node ×400 showing numerous well-formed granulomas and giant cells (courtesy Professor Peter Isaacson). (C) 18-FDG positron-emission tomogram axial image through the chest showing uptake within the mediastinal lymph nodes and the lung fields on both sides.
Around 30% of cases resolve within 2 years, particularly with single system involvement, 30% have a relapsing form and 30% progressively deteriorate.
Epidemiology
Differences in the prevalence of the disease have been known for decades; it has long been known that the incidence is highest in Nordic countries 11–24 per 105,4 and lowest in south-east Asian countries (0.85 in Korea, 2.17 in Taiwan and 1.01 in Japan).5–8 African Americans within the USA have three times the incidence of European Americans,9 and the incidence is lower still in Hispanic Americans.10 Recent studies from the Swedish registry of sarcoidosis have identified an annual incidence of 11 per 105 and a prevalence of 160 per 105,11 and in the USA three studies have shown an annual incidence of 11 per 105.12 In the UK the incidence was 14.5 per 105 between 1977 and 1983 on the Isle of Man,13 and recently 9.7 per 105 in a small study in Hertfordshire, in which the prevalence of multisystem disease at onset was seen to be 50%.14
The age of onset varies, with very few cases occurring in children and the elderly, and a mean age of onset between 45 and 55 years, later in women than men. Overall it is more common in women than in men.15
Over time the mortality rate is greater than in the general population, around 7%,16 relating predominately to the severity of the disease in those with respiratory, cardiac and neurological involvement, but also to an increased risk of infection. Patients with sarcoidosis also have more other morbidities, such as venous thromboembolism, cardiovascular disease and haematological and skin cancers,3 and a higher prevalence of other autoimmune diseases, in particular thyroid disease, connective tissue diseases and multiple sclerosis.8 16 17
Neurological involvement
Accounts of neurological complications in the disease began to be published as soon as it was recognised as a multisystem disease by Winckler in 1905,18 and by Heerford 4 years later.19 A review in 1948 found 118 published cases, in which facial and optic neuropathies were seen most commonly, although any part of the nervous system was affected.20 Later cohort studies of 285–807 patients attending systemic sarcoidosis clinics identified an incidence of neurological involvement of 3.5%–7%.19–23 More recent case series comprising 19–54 cases21–37 showed that around half of patients have isolated cranial neuropathies, of which the facial nerve is by far the most common, with peripheral nerve involvement in 10% and muscle involvement in 5%, and with brain and spinal cord lesions accounting for the remainder; 16%–70% of these series. The prevalence of neurological complications in one series from Japan was 7.2%.7
Cranial neuropathy
Facial neuropathy accounts for 70% of isolated cranial neuropathy. Most are unilateral and occur at the onset of the disease. Among those undergoing a spinal fluid examination, most have mild abnormalities of protein and cell count. Imaging is normal in half the cases, but in others shows nerve enhancement, and rarely shows more widespread changes in the absence of other clinical features.38
Optic neuropathy is common,20 38–41 arising through several different processes. A subacute optic neuritis is the most common and presents in an identical way to a demyelinating optic neuritis in most respects; pain is less prevalent, and the nadir acuity slightly less. Synchronous bilateral optic neuritis is uncommon, but sequential optic neuropathies occurred in 30%.41 Concurrent intraocular inflammation developed in 36% of one study.41 The associated field defects are central, centrocaecal or altitudinal.
An optic perineuritis occurs when the optic nerve sheath becomes inflamed, leading to visual field constriction, disc swelling and pain. Chiasmal involvement is common when a basal leptomeningitis involves the hypothalamus and adjacent structures. Finally, a compressive optic neuropathy may arise when a dural inflammatory mass, usually at the orbital apex, involves the optic nerve. There may also be sensory loss and pain.
Imaging usually shows the causative abnormality with enhancement of the inflamed area (figure 2). The cerebrospinal fluid (CSF) tends to be inactive or shows a mildly elevated CSF protein. Matched oligoclonal bands are uncommon; unmatched bands are not seen.

Cranial neuropathy in neurosarcoidosis. (A, B) Left optic neuropathy; high signal within the nerve with associated enhancement. (C) Enhancement of the right third nerve. (D) Enhancement of the left seventh nerve. (E, F) Enhancement of the left seventh and eighth nerves.
Isolated cranial neuropathies other than optic (II) and facial (VII) are less common but there are descriptions of each nerve being involved; the oculomotor (III), trochlear (IV and abducens (VI) were involved relatively frequently in previous series, the trigeminal (V) often alongside other nerves and the vestibulocochlear (VIII) proportionately less frequently.38 Imaging is usually normal, but there may be nerve enhancement.42 Inflammatory masses within the orbit, at the orbital apex and cavernous sinus may cause diplopia, trigeminal sensory loss and pain, and proptosis,40 41 and spread from an adjacent basal leptomeningitis may also involve these nerves.
Isolated hearing loss is uncommon in sarcoidosis; it occurs suddenly, often alongside a vestibular syndrome, progressively deteriorates43 44 and may fluctuate.23 Corticosteroids may rescue hearing43 but most do not recover. Imaging is either normal or shows a pachymeningeal thickening within the internal auditory canal.43 44 Spread from mastoid sinuses into the middle ear may also occur. The vestibulocochlear nerve may be involved as part of a spreading pachymeningitis or a leptomeningitis in the region, often with accompanying facial nerve palsy.
Isolated neuropathies of the lowest cranial nerves are uncommon, but I have often seen a bulbar disorder characterised by progressive dysphonia and dysphagia, although this is infrequent in the literature.20 Patients in my series were all female, and most had normal imaging but slightly active CSF.38 Occasionally there is weakness and atrophy of one side of the tongue.20 Again, involvement of these nerves may form part of a more widespread disorder due to a basal meningitis.
Peripheral neuropathy
Peripheral neuropathy accounts for 10%–14% of some series, but much less in others. Reports citing a high prevalence also note concomitant central neurological disorders and do not provide neurophysiological proof of peripheral involvement. My own view is that peripheral neuropathy is uncommon in sarcoidosis, accounting for <5% of all cases, and is not associated with concomitant central neurological disease. It comprised only 0.3% of 3475 nerve biopsies carried out in a tertiary centre in Paris.45
The symptoms are sensorimotor or purely sensory, and the electrophysiological investigations point to an axonal pathology predominately, although there may be conduction slowing, focal conduction block and multifocal conduction block.46 47 Mononeuritis multiplex and an asymmetric neuropathy may occur.45 46 48 49 Mononeuropathies, particularly of the radial and ulnar nerves45 48 49 can occur; in one case50 there was a granulomatous inflammatory mass in Guyon’s canal.
An acute inflammatory demyelinating polyradiculoneuropathy is rare and seems to arise at the onset of the systemic disease; the electrophysiological findings are typical, but the CSF is active and includes a lymphocytosis. Corticosteroids appear to resolve the disorder.45 48 51 52 The neuropathology is of a granulomatous infiltration of the epineurium with a perivascular infiltration of inflammatory cells and granulomas around the nerve (figure 3). There may be nerve fibre disorganisation and axonal loss.45 46 53 54 Pathological features of a perineural vasculitis occurred in 40% of Professor Said’s cases. Other authors have also noted that the neuropathy develops years after the onset of the systemic features.
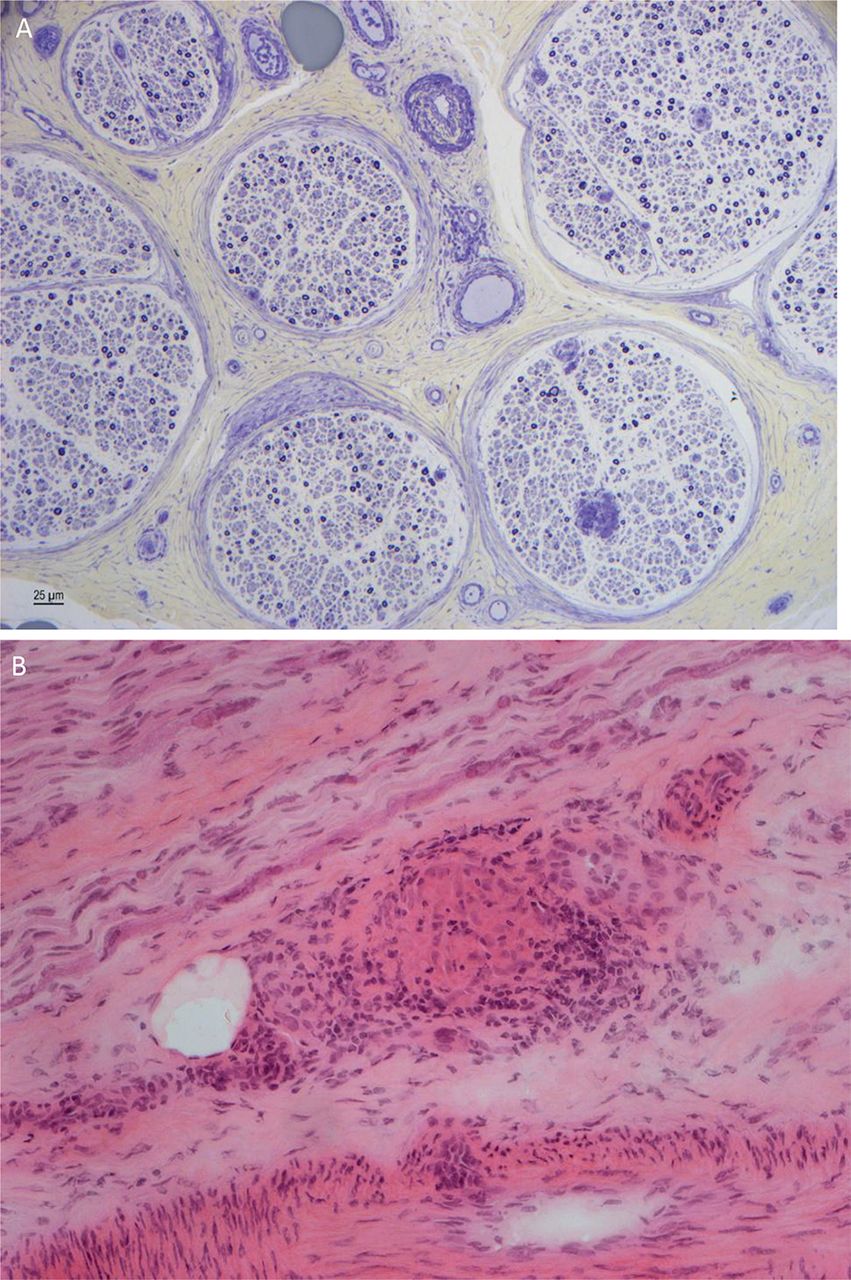
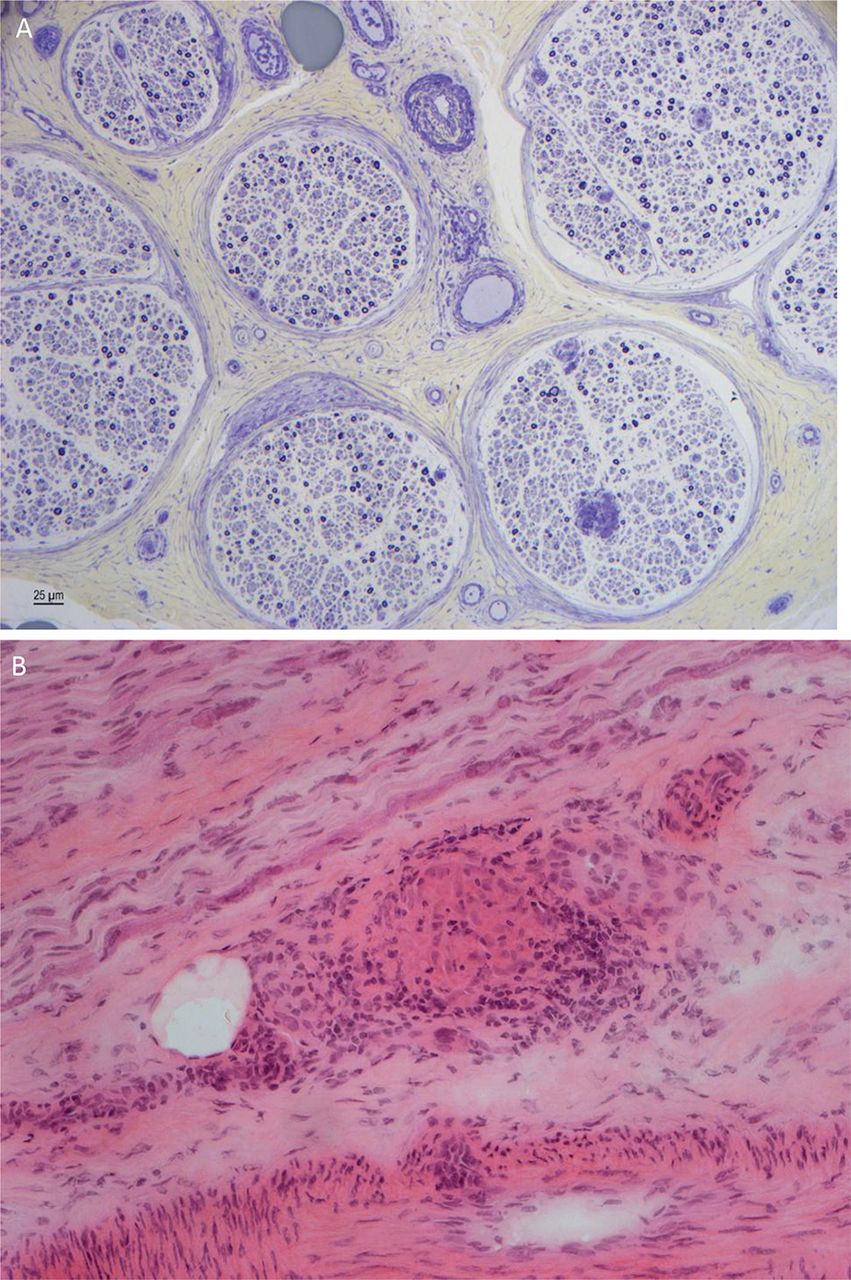
Sural nerve biopsy showing depletion of myelinated fibres, axonal degeneration and regenerative clusters (A). There is a perivascular cellular infiltrate with T and B lymphocytes and giant cells, and a granuloma (B) (courtesy Dr Rosalind King).
There has not been a trial of treatment. There are reports of a response to intravenous immunoglobulin in those with a multifocal or demyelinating disorder,52 yet another was unresponsive to intravenous immunoglobulin who subsequently improved with corticosteroids.55 My own treatment paradigm is to treat those with inflammatory neuropathies with intravenous immunoglobulin while also treating the systemic disease with immunosuppression, and managing those with an axonal pathology with treatment appropriate to the systemic disease alone; in my experience these patients tend not to deteriorate over time.
Small fibre neuropathy is also uncommon but associated with a severe and very poorly treatment-responsive distal neuropathic pain. Large fibre nerve conduction studies are normal, thermal threshold and cutaneous autonomic responses subnormal, and intraepidermal nerve fibre density reduced in all cases.56 A series of 115 patients also had a high prevalence of accompanying autonomic symptoms (53%) such as cardiovascular instability, disorders of sweating and gastrointestinal transit delay.57 Intravenous immunoglobulin,57 tumour necrosis factor alpha (TNFα) blockers58 and the erythropoietin derivative ARA-29059 can help the symptoms, but there has been no formal comparative trial of treatment; the disorder does not resolve. The pathogenesis is unknown, since there is no granulomatous inflammation within the nerves, but may relate to high circulating concentrations of chemokines, which induce hyperexcitability within fibres leading to the appreciation of pain; calcium influx induces an oxidative stress leading to axonal degeneration.
Some patients, at the onset of the systemic disease, present with a burning numbness of the chest wall that responds to corticosteroids and, in some cases, has been relapsing and corticosteroid-dependent for a time. Imaging of the spinal cord has been normal. Thoracic radiculopathy appears to be exclusive to sarcoidosis and is noted in early reports. Active CSF despite normal imaging suggests a meningitis of the dorsal spinal cord, picking off the roots as they leave to form intercostal nerves. Other cases60 61 have prolonged F wave responses, compatible with this assumption. The rapid resolution with corticosteroids and yet a propensity to relapse on withdrawal suggests that the nerve roots are inflamed; indeed, a recent report showed thoracic root enhancement in a patient with an adjacent paravertebral inflammatory mass.62
Pituitary and hypothalamic involvement
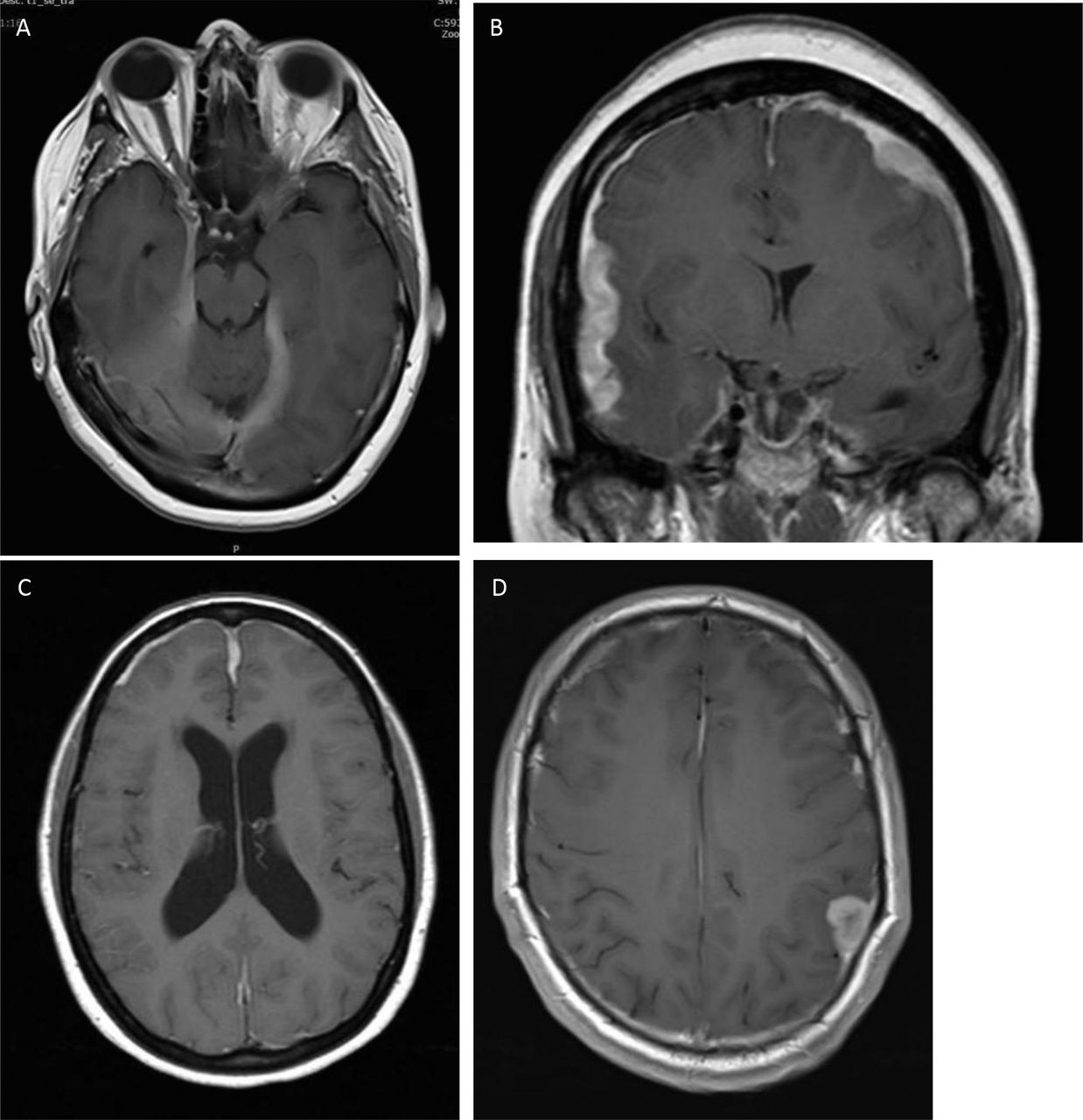
Hypothalamic involvement, in particular associated with diabetes insipidus, has featured in the literature from the earliest reports. Most present with endocrine symptoms, in particular polydipsia.63 64 Most patients present with a gradual panhypopituitarism while others have endocrine involvement as part of an increasing leptomeningitis. Other presentations include headache, cranial neuropathies causing diplopia and facial sensory loss and of course visual symptoms caused by optic chiasm involvement. In my series of six cases, three had isolated pituitary involvement and in three the disease had spread to the adjacent cavernous sinus, causing third and sixth cranial neuropathies. Imaging was abnormal in all but one (figure 4). Those with hypothalamic involvement all had additional more widespread leptomeningeal disease and therefore other symptoms and signs. Two of these had enlargement of the stalk but not the gland; both had diabetes insipidus.

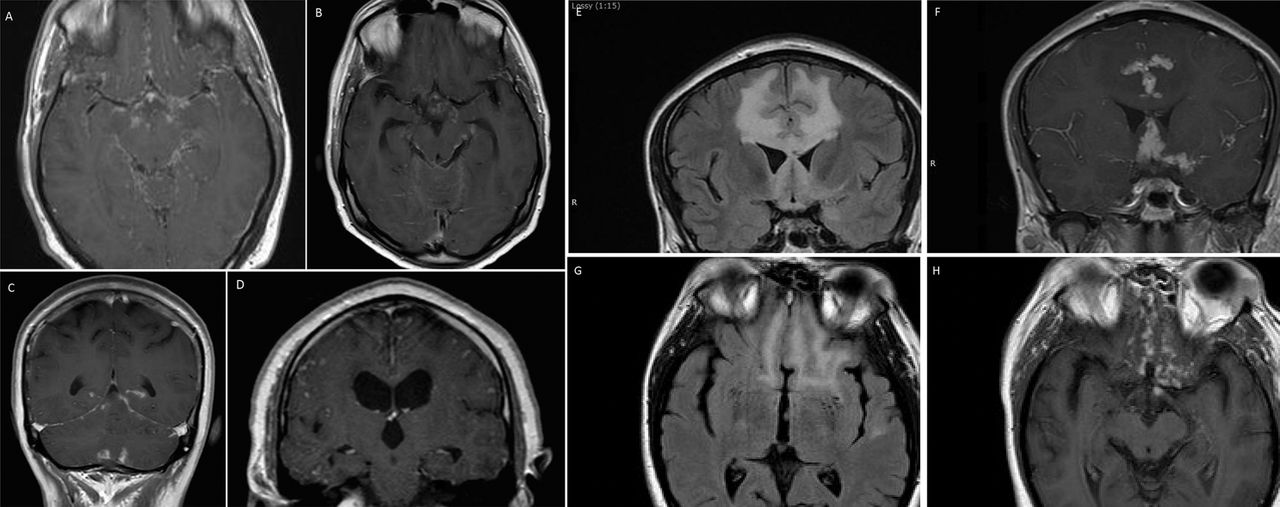
Involvement of the pituitary and hypothalamus. (A) Isolated enlargement of the anterior pituitary presenting with headache and visual loss. (B) Anterior pituitary mass with adjacent leptomeningeal involvement extending into the cavernous sinuses on both sides. The patient presented with diplopia and was found to have asymptomatic hypopituitarism. (C) Widespread nodular leptomeningeal disease with enlargement of the anterior and posterior pituitary extending into the hypothalamus. (D) More widespread leptomeningeal disease most prominent within the hypothalamus. (E) Leptomeningeal disease of the brainstem, optic chiasm and hypothalamus.
Endocrine investigations mostly identify gonadotropin and thyrotropin deficiency, diabetes insipidus in over half and corticoadrenal insufficiency in the least.63 64 Most do not improve with treatment despite a radiological response, although some recover completely.63
Brain involvement
Granulomatous inflammation may involve the central nervous system through three processes: a pachymeningitis, a leptomeningitis and a vasculitis (figure 5).

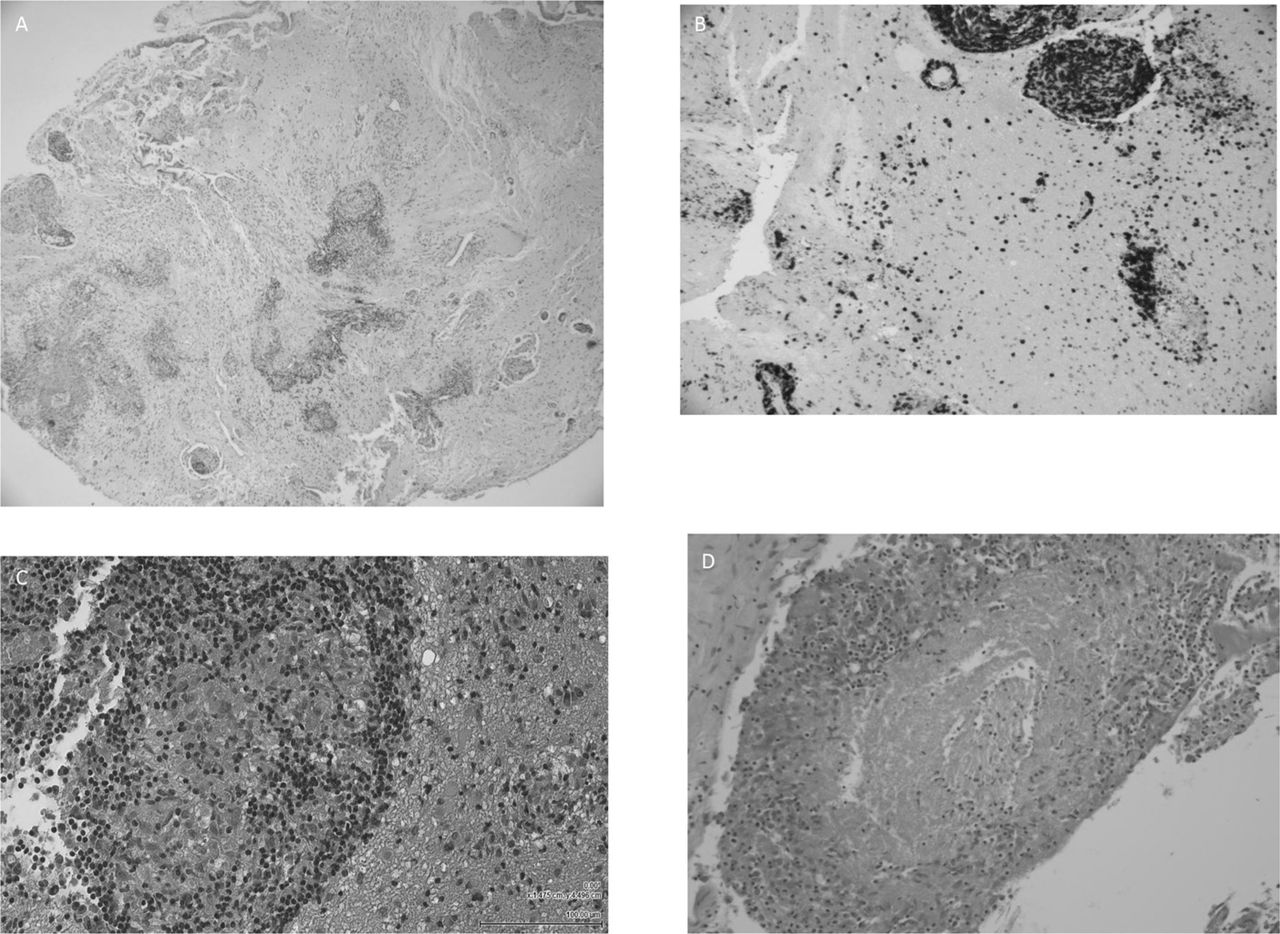
Neuropathology of neurosarcoidosis: H&E-stained sections (A, C, D) of brain (A, C) and leptomeninges (D) showing a prominent infiltration of small well-formed epithelioid granulomas surrounded by lymphocytes (B) and plasma cells (courtesy Dr Malcolm Galloway).
Pachymeningitis
Dural inflammation may develop in any part of the cranial cavity; the basal regions and the convexity. Around half present as mass lesions with focal neurological signs, often with seizures. The remainder have a multifocal en plaque type appearance that may be very widespread.38 Headache is common, and no one in this patient group in my series had hydrocephalus. When the cavernous sinus and orbital apex were involved, patients presented with pain, diplopia and optic neuropathy. The CSF was active in most but this did not correlate with clinical features or severity.38 Imaging was abnormal in every case (figure 6).
Pachymeningeal disease in neurosarcoidosis. Each patient presented with headache and focal neurological signs. Patient D had had a seizure.
Patients thus present with headache and focal neurological signs and are less impaired than those with a leptomeningitis. Following treatment, which is required for several years, the outcome is good.65
Leptomeningitis
75% of cases in the Royal Free series38 showed features of an invasive and destructive meningoencephalitis. Two-thirds presented with signs of diencephalic dysfunction and hydrocephalus, while a third had signs of involvement of the brainstem, with associated hydrocephalus. The basal meninges were more commonly involved than the convexities. The disease course was subacute but increasingly rapid, with rapid accrual of a severe encephalopathy and neurological signs. These patients are more disabled than those with a pachymeningitis,65 and the two processes do not seem to overlap.
Imaging was abnormal in every case (figure 7). The CSF was active in most cases, with a high protein concentration and cell count and low CSF/serum glucose ratios. CSF protein concentration correlated with the presence of hydrocephalus.

Leptomeningeal disease in neurosarcoidosis. (A) Widespread basal leptomeningeal enhancement involving the diencephalon and brainstem. (B, C) More localised nodular leptomeningeal enhancement of the brainstem and the cerebellum. (D) Widespread enhancement extending into the brain causing a subacute encephalopathy with headache. (E, F, G, H) A severe destructive invasive leptomeningitis of the frontal and temporal regions leading in both cases to impairments of cognitive function, motor control and seizures.
Treatment should be intensive; corticosteroids alone will not be sufficient and may lead to repeated relapse leading to irreversible neurological impairment. The outcome depends on the timing and type of treatment (figure 8).
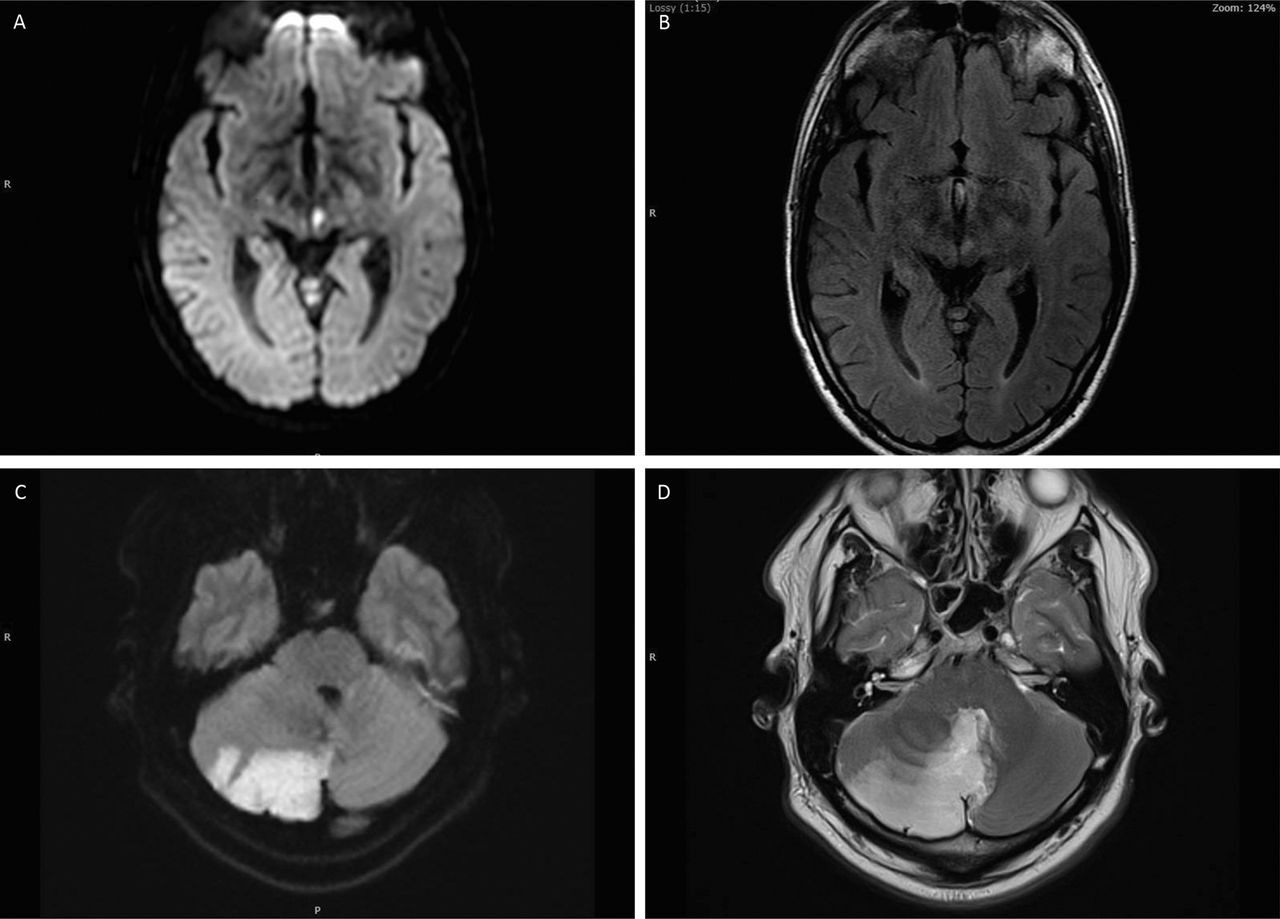
Vascular involvement in neurosarcoidosis. (A, B) Infarction of the midbrain. (C, D) and cerebellum in patients with new onset systemic sarcoidosis and no risk factors for early onset cerebrovascular disease. In each case vascular and cardiac imaging was normal.
Vascular involvement
Early pathological descriptions showed that the predominate pathological process was a granulomatous inflammation within the leptomeninges, spreading into the subjacent parenchyma.66–68 Occasionally the walls of small and medium-sized arteries show epithelioid cell invasion, leading to an inflammatory response with disrupted internal elastic lamina and fibrosis, together leading to occlusion or stenosis of the vessel lumen.66 However, only one report noted infarction of the adjacent parenchyma.68
Clearly, therefore, vascular involvement does occur but the small number of case reports available implies that it rarely has a direct clinical consequence; such reports include infarction due to large vessel occlusion occurring without prelude,69–72 while others note a crescendo series of transient ischaemic attacks culminating in infarction.72–74 Half these cases had an adjacent leptomeningeal enhancement while the others showed only vascular changes on imaging (figure 9).69–71 74 Antiplatelet therapy does not reduce the frequency of attacks,73 74 whereas corticosteroids do.72 74

Enhancement of parenchymal vessels in neurosarcoidosis.
Arteriography may be normal,69 70 74 or show focal stenosis of the anterior cerebral artery, middle cerebral artery and internal carotid artery; abnormalities resembling moyamoya may develop.71–73 75 76 A recent case showed that this probably relates to a large vessel arteritis.77 Another series has confirmed that vessel wall enhancement related to stroke in more than half the cases compared with 18% of those with inflammation and no radiological signs of infarction (figure 9).78
Intracranial haemorrhage is also rare,79 probably as uncommon as cerebral infarction. Patients all have clear concurrent evidence for a granulomatous leptomeningitis. The haemorrhage may be subarachnoid but more commonly is lobar. They may be multiple,80 infratentorial79 and in one case within the spinal cord.81 A neuropathological examination showed inflammation within and disruption of the integrity of the walls of small veins, suggesting an inflammation of the wall leading to leakage at low pressure.79 Recent reports of vein wall enhancement and venous engorgement in the medullary veins on vessel wall imaging82 and susceptibility-weighted MRI83 support this hypothesis.
Spinal cord and cauda equina involvement
Involvement of the whole cranium and spinal canal is common both in leptomeningeal and pachymeningeal forms of the condition, but isolated disease of the spinal cord and cauda equina does occur. Most present with a subacute spinal cord lesion, in which there is a single cervical or thoracic longitudinally extensive lesion, associated with adjacent leptomeningeal enhancement (figure 10A–D).38 84–88 Pachymeningeal lesions tend to be smaller and present as compressive lesions. Half of those with lower dorsal lesions also have cauda equina involvement, with nodules of enhancement on the descending roots (figure 10E,F).38 89 Individual roots may also be affected. The signs of a cauda equina lesion involve early sphincter disturbance and a predominately sensory disorder; it is rarely painful. However, a motor disorder comprising a single motor root usually is painful.

T2-weighted and T1-weighted MRI following contrast administration showing (A, B) a lesion of the cervical and (C, D) thoracic cord and (E, F) cauda equina showing enhancement of inflamed nerve roots.
A progressive cord lesion is less common; in this situation imaging tends to be normal or showing signs of atrophy38; when there is an accompanying amyotrophy due to root involvement, this may be difficult to distinguish from motor neuron disease, but the CSF tends to show active cells.
Isolated neurosarcoidosis
Patients may present with a neurological disorder characterised by granulomatous inflammation with no clear signs on imaging of a systemic disease. Often patients present with the neurological disorder and signs of the systemic disease are identified by search; in some no evidence is found, in others the systemic features develop only later.90 The imaging features and CSF abnormalities are similar to those with systemic disease as well,38 except that only those with isolated neurosarcoidosis have unmatched oligoclonal bands. A series of 10 patients from two US centres found no difference in clinical, imaging or CSF features of the disease.91 A comprehensive search for a masked infection or tumour is essential in these patients.
Imaging
MRI is the most important imaging modality; in one recent series38 it was abnormal in 35% of those with cranial neuropathy other than optic neuropathy, and 100% of those with central neurological disease. It is essential to give a paramagnetic contrast agent, since contrast enhancement is often the only abnormality, and only a small minority show no enhancement. As noted above, meningeal enhancement correlates with the site of the disease, and in leptomeningeal disease enhancement and oedema of the subjacent cortex and white matter is common, and some patients show enhancement of the affected vessel walls.77 78 82 Patients with progressive spinal cord disease may have no abnormalities or atrophy, and those with vascular involvement as noted above, may only have abnormalities of perfusion, diffusion or magnetic susceptibility. Positron emission tomography (PET) studies with 18-FDG92 93 may show abnormalities when postcontrast MRI does not.
Cerebrospinal fluid
The CSF is always active in untreated neurosarcoidosis. The protein is raised, there is a lymphocytosis, and the CSF/plasma glucose ratio is reduced. In a recent series of 128 patients with neurosarcoidosis affecting the brain, spinal cord and cauda equina,38 the median CSF protein was 0.8 (0.19–8.35) g/L, and raised in 76% of 89 samples. The median CSF white cell count was 5 (0–395) cells/µL, and raised in 51% of samples. The CSF to plasma glucose ratio was reduced in 81%, median 48%.20–79 94 95 Patients with isolated cranial neuropathies had less active CSF than those with more widespread disease, in whom CSF protein correlated both with the presence of hydrocephalus, and with disability as measured by the modified Rankin score. Those with cauda equina involvement had a higher CSF protein than those with isolated spinal cord involvement. CSF cell count and glucose ratio did not correlate with disability.
Oligoclonal bands were negative in serum and CSF in 73% of this series, and there were matched bands in 23%. The presence of matched bands correlated with CSF protein, implying a relationship with disease severity. Unmatched oligoclonal bands occurred only in the small group with isolated neurological involvement. In another series96 patients with MRI-visible leptomeningitis had higher CSF cell counts and protein concentrations, and a lower glucose concentration, than those without enhancement.
In those with peripheral neuropathy the CSF protein is modestly raised, with matched oligoclonal bands but without cells. In my series of small fibre neuropathy the CSF was inactive but there are no data from other studies.
Serum biomarkers
At the onset of the systemic disease a peripheral lymphopenia is common (due to accumulation of activated T cells at the sites of inflammation). There is also a cytokine and chemokine pattern in the serum, although this varies with the clinical syndrome and the disease time course.97 IgG deficiency may also develop.
Raised serum calcium (due to upregulation by granulomas of an enzyme that converts 25-hydroxy vitamin D to 1,25 dihydroxy vitamin D) occurs in 10% of cases, and hypercalciuria in a further 40%.
Angiotensin-converting enzyme (ACE) is secreted by alveolar granulomas and 60% of patients with systemic sarcoidosis have a raised serum ACE concentration . It is not useful as a disease monitor since it does not always rise again in relapse and does not define disease progression in lung disease.98 It is also raised in numerous infections, including tuberculosis, other inflammatory illnesses and in diabetes mellitus, hypothyroidism and lymphoma.
Serum amyloid A1 is also released by activated macrophages and may be raised when the serum C-reactive protein is not. It regulates granuloma formation through toll-like receptor 2. Lysozyme, neopterin and chitotriosidase are also released by macrophages and are raised in blood and in broncho-alveolar lavage fluid,98 but none is specific for sarcoidosis.
A recent comparative study of ACE, chitotriosidase, lysozyme and KL-6 found that KL-6 is associated with lung fibrosis and that a raised chitotriosidase had a specificity of 85% in sarcoidosis.99
CSF biomarkers
The spinal fluid assay results from a large series are summarised above. The CD4/CD8 ratio is raised in CSF (and broncho-alveolar lavage fluid) in people with neurosarcoidosis; in one study,100 the interleukin 6 (IL-6) and IL-10 concentrations were raised, more than in multiple sclerosis, and more in patients with active disease than in those previously treated. CSF cytokines are raised in all inflammatory and infective neurological diseases, and we need further work to define the relevance of these features in the diagnosis of neurosarcoidosis. CSF ACE is not a biomarker; ACE increases in proportion to CSF protein and is raised in any inflammatory or infective disease in which the protein is raised.
Differential diagnosis
The need for a histologically sound basis for the diagnosis is increasing as specific biological therapies are developed. The differential diagnosis is complex and wide ranging; neoplastic disorders such as lymphoma, meningioma, glioma, choristomas and metastatic carcinomas may all cause a clinical syndrome with radiological abnormalities consistent with the disease, and indeed granulomatous inflammation may develop adjacent to a neoplastic lesion, particularly lymphoma and germinoma. Infectious diseases due to Mycobacterium tuberculosis, M. leprae, spirochaetae, brucella, bartonella, toxoplasma and various parasites and fungi may also do so. Other granulomatous diseases that affect the nervous system include primary angiitis of the central nervous system and idiopathic hypertrophic pachymeningitis. Lymphocytic disorders may give similar imaging and clinical appearances, including chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS), the recently described auto-immune glial fibrillar acidic protein astrocytopathy, the neurological complications of systemic lupus erythematosus, Sjögren’s syndrome and rheumatoid arthritis, neuromyelitis optica spectrum disorder, IgG4-related disease and cryopyrin-associated periodic syndrome. Histiocytic diseases such as Langerhans cell histiocytosis, Erdheim-Chester disease and Rosai-Dorfman syndrome may also cause disorders with identical imaging to neurosarcoidosis. Systemic and neurological granulomatous disease may also arise in inherited immunodeficiency disorders and as part of an immune reconstitution syndrome in acquired immunodeficiency. Sarcoid-like reactions may also arise during immune treatment of neoplastic and inflammatory diseases such as rheumatoid arthritis (with TNFα and IL-6 blockers), multiple sclerosis (with interferon β, daclizumab and alemtuzumab) and during cancer treatment with immune checkpoint inhibitors.
Patients with systemic sarcoidosis have an increased prevalence of certain neurological infections, in particular cryptococcus, listeria and progressive multifocal leukoencephalopathy; a brain lesion in sarcoidosis does not automatically reflect spread of the inflammatory condition into the nervous system. Finally, as noted above, patients with sarcoidosis are at increased risk of developing other inflammatory diseases, including multiple sclerosis, undoubtedly owing to their genetic susceptibility to develop other immunological diseases.
Diagnosis and diagnostic criteria
I am surprised still to learn of patients diagnosed with neurosarcoidosis on the basis of having ruled out multiple sclerosis and neuromyelitis optica spectrum diseases, the criterion seemingly being, ‘What else might it be?’. This is particularly likely when the neurological disorder is associated with inflammatory ocular disease, which may complicate many other inflammatory, infective and neoplastic diseases, and which not even be associated with the neurological disease of sarcoidosis. There is a large list of conditions that can simulate the clinical and imaging characteristics of neurosarcoidosis, and it is very important to understand that a neoplastic and infective diseases can initiate a granulomatous disease.101 All of this makes it virtually impossible to create meaningful and specific diagnostic criteria. The concept of definite, probable and possible neurosarcoidosis is understandable, but it can readily be seen that patients with biopsy-proven granulomatous disease of the central nervous system (CNS) may not have sarcoidosis, and that it is impossible to diagnose possible neurosarcoidosis with any certainty whatsoever, given that there is no evidence for sarcoidosis systemically and no confirmation that the neurological disorder is granulomatous,. More recently the World Association of Sarcoidosis and Other Granulomas Diseases102 has published a carefully considered organ assessment instrument that will be much more useful to clinical trials and research than the previously published diagnostic criteria. In addition, a consortium of American experts has compiled a clearly considered summary of the investigation of the disease and notes ‘one can never be 100% certain of the diagnosis of sarcoidosis, even with a brain biopsy …’,103 and provides a further proposal for diagnostic criteria, disappointingly although understandably continuing with the theme of definite, probable and possible forms of the disease.
Over time genetic and biochemical markers will replace any clinical criterion, and we can only hope that this occurs soon.
Treatment and outcome
Neurosarcoidosis is a treatable disease and its management in a correct and timely way can avoid patients having severe residual neurological impairments.65 Corticosteroids form the basis of treatment in all granulomatous diseases; the inflammation is corticosteroid-responsive but relapses readily if treatment is reduced too quickly. Some require very high doses (even 100 mg/day) to maintain a treatment response; over time the disease ameliorates, and the dose may reduce without relapse. Immunosuppressive agents are essential not only as steroid-sparing but as disease modifiers; corticosteroids reduce the inflammation, but the immunosuppressive agent prevents it from returning. Methotrexate, mycophenolate mofetil and azathioprine are all used; methotrexate is better than mycophenolate mofetil in systemic104 and neurological disease,105 and I have not found azathioprine sufficiently effective in central neurological disease. The standard dose of methotrexate is 15–20 mg weekly with folate rescue, with the dose altered according to the effect on the disease, the corticosteroid dose, and the prevalence of adverse effects. Intravenous cyclophosphamide may be used for severe disease; it has more adverse effects including serious infections but is associated with reduced relapse frequency.106
The biological agent infliximab, which blocks TNFα receptors through binding to TNFα, is effective in the systemic and ocular forms of the disease107 108 and in those with neurological involvement.65 109 110 Its use has substantially improved the outcome of treatment in severe forms of the disease, particularly invasive leptomeningeal disease and treatment-resistant pachymeningeal disease,65 and may over time remove the disease entirely.65 However, this takes 2–5 years of continual treatment. Incomplete treatment risks early relapse following treatment withdrawal108 109 111–113; in one series this was 50% in patients for whom treatment was stopped after a year, but when treatment continues until meningeal enhancement resolves (over 18–36 months) the risk of relapse was less than 10%.65 Use of biosimilars is currently being studied; in one series111 patients starting with a biosimilar seemed to respond as well as those treated with originator (although both groups relapsed). However, it may be hazardous to switch from originator to biosimilar during treatment,111 including recurrence of leptomeningeal enhancement and the development of hydrocephalus.114 It may be safe to use biosimilars from treatment onset; further study is required to define this and to ascertain whether or not the response is the same. Other TNFα blockers have less effect; adalimumab is slower to take effect, and golimumab and etanercept have not shown an effect in respiratory involvement,115 and etanercept can cause sarcoidosis in patients who receive this to treat connective tissue diseases.
There are isolated reports of a response to the IL-6 blocker tocilizumab,100 116 and the CD-20 antagonist rituximab117 but the IL-17 blocker ustekinumab does not help the respiratory disease.118 Since janus kinase/signal transducers and activators of transcription (JAK-STAT) signalling may occur in the pathogenesis of sarcoidosis, Janus kinase inhibitors may be beneficial119; there are recent reports of an effect in cutaneous and respiratory disease.120 Inhibitors of mammalian target of rapamycin complex 1 (mTORC1) may also play a role in the future.121
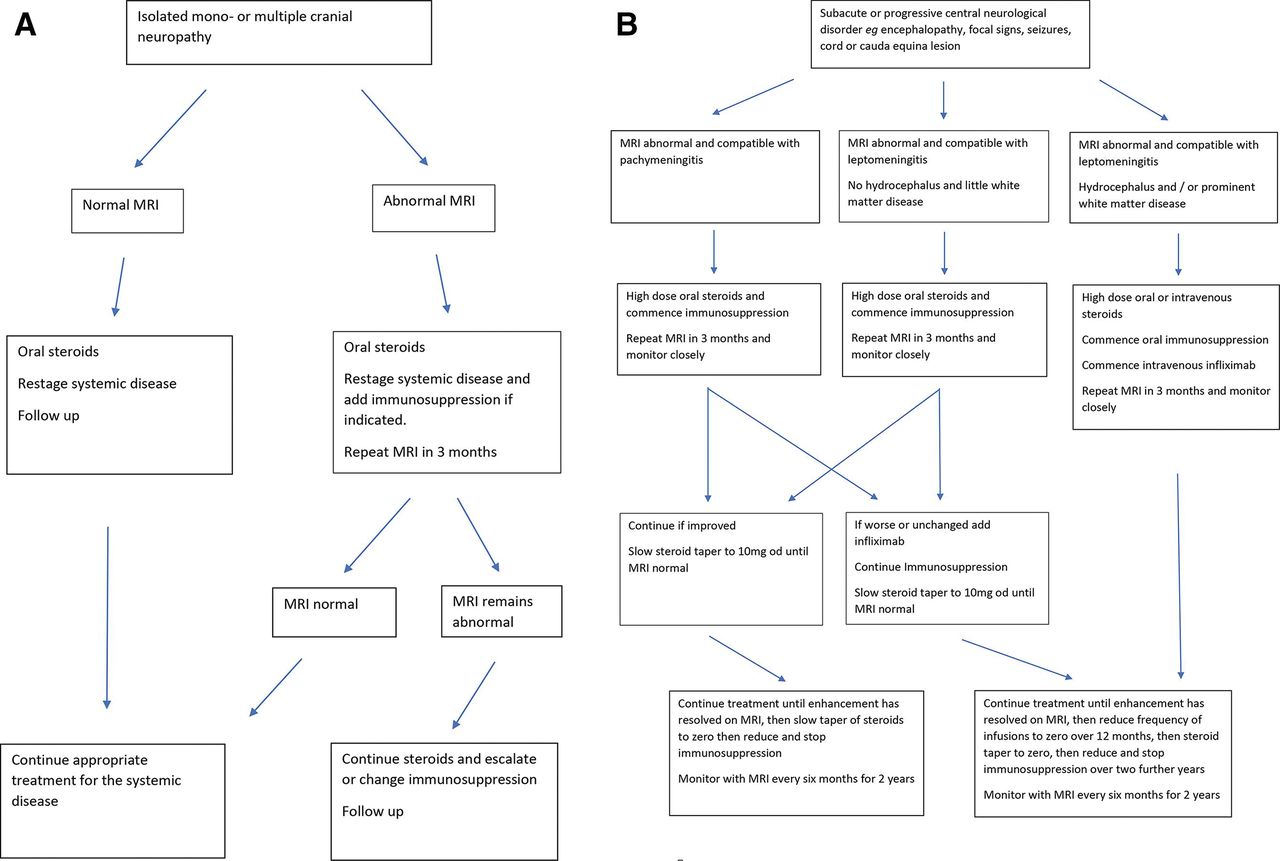
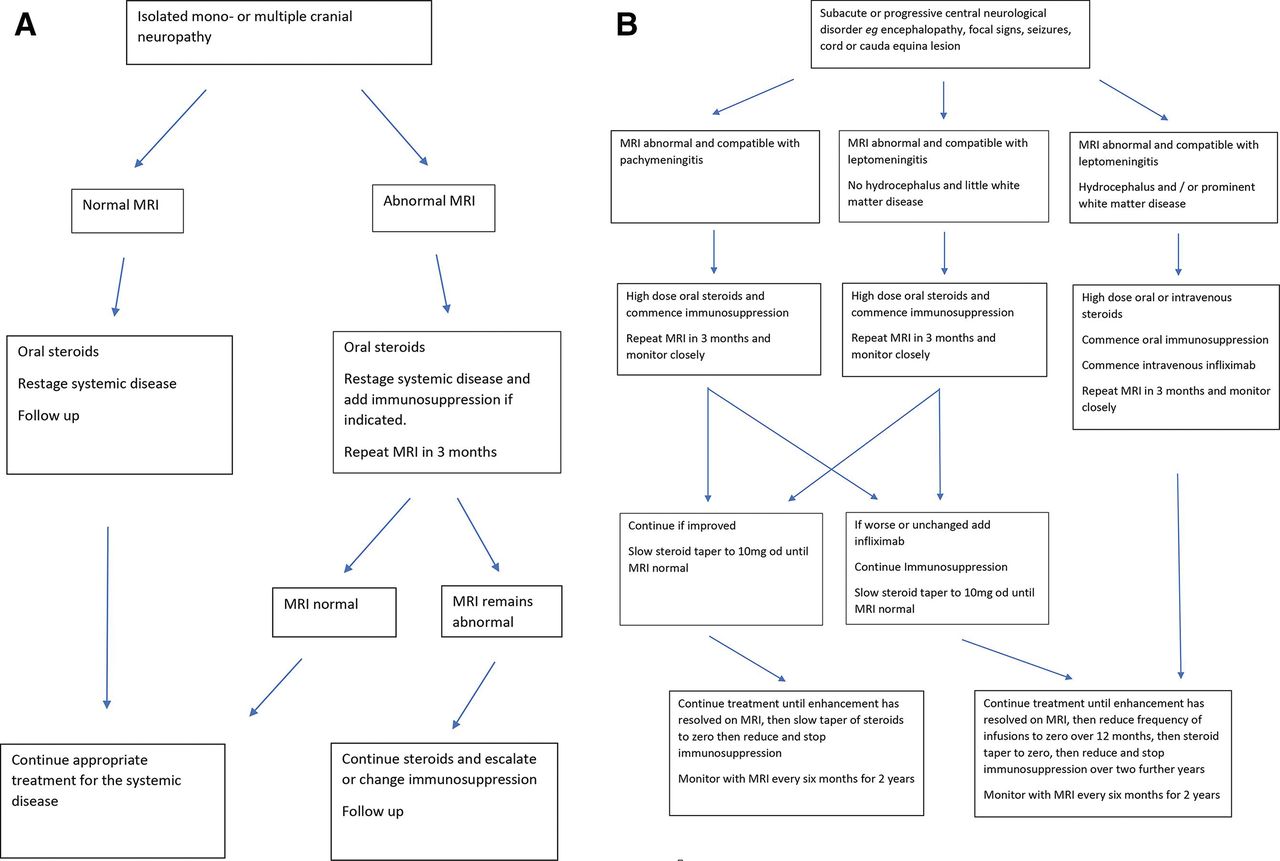
Perhaps 20% of patients with neurosarcoidosis need biological therapies65; those with cranial neuropathy and peripheral neuropathy respond to an increase in treatment for the systemic disease, and this is usually sufficient; there is only a small risk of relapse or of further symptom, and there is no evidence that patients with peripheral nerve involvement have a coexisting central neurological disease. Most patients with a mild central disease of the brain or spinal cord respond to oral corticosteroids and immunosuppression. Those with rapid onset disease in whom it is clear that the disease is destructive and deteriorating should be treated urgently with biological therapies. Figure 11 shows a treatment paradigm developed in our unit and based on experience of treatment of 235 cases.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A suggested treatment algorithm for neurosarcoidosis causing (A) cranial neuropathy and (B) involvement of the brain and spinal cord by pachymeningitis and leptomeningitis.
Further reading
Grunewald J, Grutters JC, Arkema EV, et al. Sarcoidosis. Nat Rev Dis Primers 2019;5:45.
Kidd DP. Sarcoidosis of the central nervous system: clinical features, imaging and CSF results. J Neurol 2018;265:1906-15.
Key points
Neurosarcoidosis is uncommon, occurring in 5% of those with the systemic disease and 2 per 100 000 of the general population.
Seventy per cent have a mild disease comprising cranial neuropathy, radiculopathy and peripheral small-fibre or large-fibre neuropathy.
Thirty per cent have a severe CNS inflammatory disease requiring corticosteroids and immunosuppression; those with severe disease at onset or those who are treatment unresponsive should receive biological therapies, including TNFα antagonists.
References
Footnotes
Contributors DPK wrote this review and was responsible for the interpretation of the data presented and the cited literature.
Funding The author has not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Other content recommended for you
- Immune-mediated conditions affecting the brain, eye and ear (BEE syndromes)
- Chronic meningitis, seizures and myoclonus
- A practical approach to the diagnosis of spinal cord lesions
- Neurosarcoidosis associated with intracerebral haemorrhage: a challenge in diagnosis and management
- Rapidly progressive dementia and ataxia in an elderly man
- Infectious encephalitis: mimics and chameleons
- Measurement of cerebrospinal fluid ACE level in aseptic meningitis: diagnostic?
- The neurology of Sjögren's syndrome and the rheumatology of peripheral neuropathy and myelitis
- Pragmatic guide to peripheral nerve disease and the role of clinical biomarkers
- Sarcoidosis of the nervous system