Article Text

Abstract
Autoantibodies to leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein like-2 (CASPR2) are associated with clinically distinctive syndromes that are highly immunotherapy responsive, such as limbic encephalitis, faciobrachial dystonic seizures, Morvan’s syndrome and neuromyotonia. These autoantibodies target surface-exposed domains of LGI1 or CASPR2, and appear to be directly pathogenic. In contrast, voltage-gated potassium channel (VGKC) antibodies that lack LGI1 or CASPR2 reactivities (‘double-negative’) are common in healthy controls and have no consistent associations with distinct syndromes. These antibodies target intracellular epitopes and lack pathogenic potential. Moreover, the clinically important LGI1 and CASPR2 antibodies comprise only ~15% of VGKC-positive results, meaning that most VGKC-antibody positive results mislead rather than help. Further, initial VGKC testing misses some cases that have LGI1 and CASPR2 antibodies. These collective observations confirm that laboratories should stop testing for VGKC antibodies and instead, test only for LGI1 and CASPR2 antibodies. This change in practice will lead to significant patient benefit.
- Autoimmune
- NMDA receptor
- Epilepsy
- Neuroimmunology
- Movement disorders
- Assay
- Autoantibody
- Neuromyelitis optica
- Encephalitis
- Neuromyotonia
- Immunology
- Paraneoplastic
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
- Autoimmune
- NMDA receptor
- Epilepsy
- Neuroimmunology
- Movement disorders
- Assay
- Autoantibody
- Neuromyelitis optica
- Encephalitis
- Neuromyotonia
- Immunology
- Paraneoplastic
VGKC ANTIBODIES: ORIGINS OF A DIAGNOSTIC TEST
The initial clinical insights describing an autoimmune basis for acquired neuromyotonia (Isaacs’ syndrome), a form of peripheral nerve hyperexcitability, emerged in Oxford around 30 years ago. A patient with severe disease, refractory to sodium channel blocking medications, showed almost complete symptom resolution after plasma exchange.1 Moreover, this patient’s purified IgG induced muscle hyperexcitability in phrenic nerve–diaphragm preparations. These findings suggested an underlying autoantibody-driven mechanism for neuromyotonia, with voltage-gated potassium channels (VGKCs) considered a likely antigenic target.
This prediction was directly tested with alpha-dendrotoxin (α-DTX), a neurotoxin derived from green mamba snake venom. α-DTX labels Kv1.1, 1.2 and 1.6 potassium channels and was radioiodinated to label soluble mammalian brain extracts upon establishment of the VGKC antibody radioimmunoassay. This radioimmunoassay detected serum or cerebrospinal fluid autoantibodies that precipitated iodinated α-DTX. Hence, these autoantibodies were originally thought to bind VGKCs,2 3 and some reports even showed IgG from patients binding directly to oocyte or HEK293T-cell-expressed VGKCs.2–4 During this series of molecular observations, VGKC antibodies were also identified in patients with Morvan’s syndrome (neuromyotonia with characteristic central nervous system manifestations)4 and limbic encephalitis.5–7 Importantly, patients with these syndromes improved markedly following immunotherapy. Overall, these serological discoveries helped to describe and classify a potentially reversible set of autoimmune neurological conditions.
Subsequently, these findings were rapidly disseminated through the neurology community. To avoid missing a potentially immunotherapy-responsive condition, VGKC antibodies were requested in patients with a wider spectrum of clinical features. This led to a large number of positive VGKC antibody results, in broad-ranging phenotypes. Many of these syndromes were not immunotherapy-responsive or intuitively immune-mediated. This non-distinctive set of syndromes led us to question how antibodies to a single protein family could cause such a variety of diseases (Figure 1).

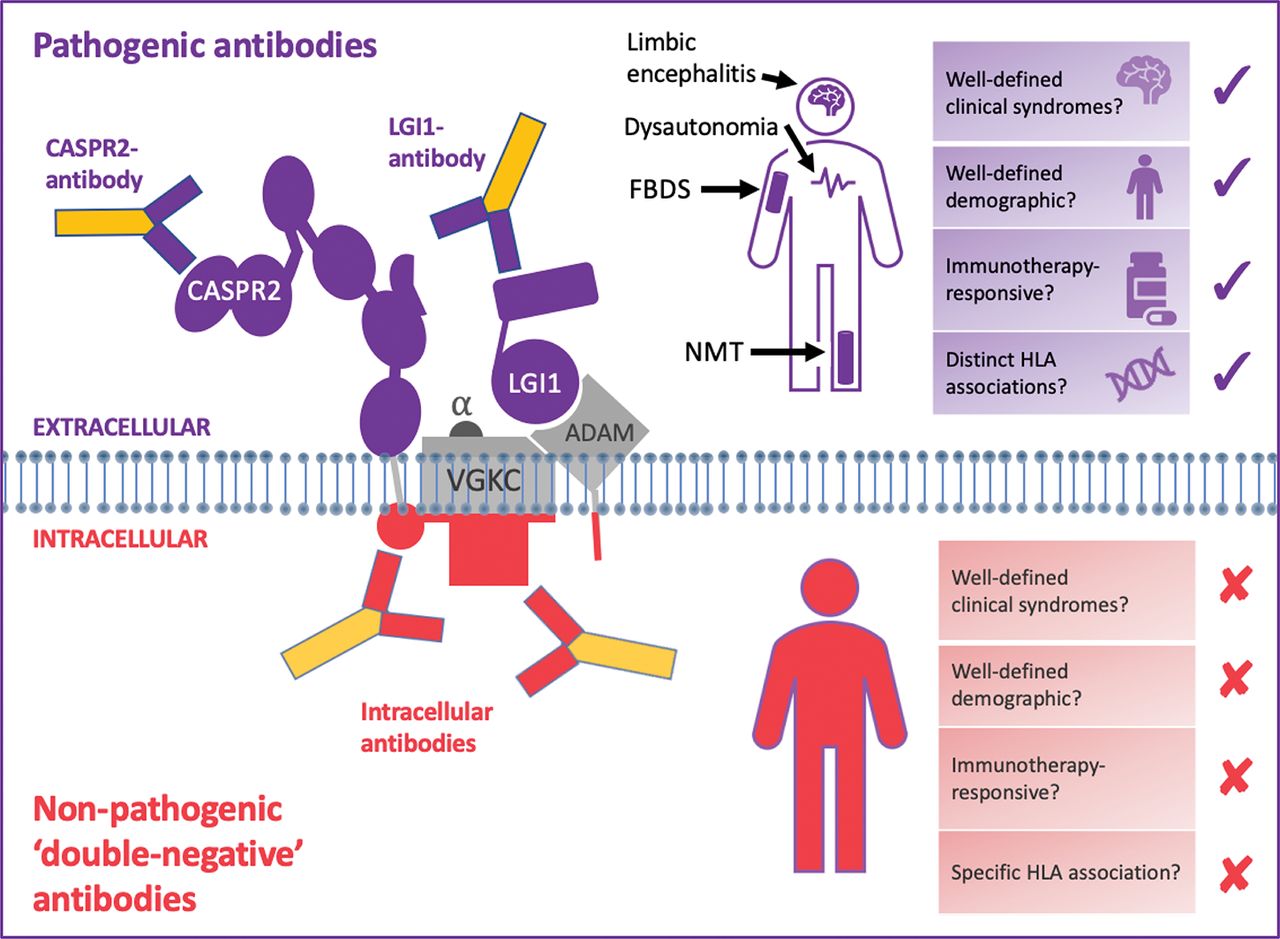
LGI1 and CASPR2—but not double-negative VGKC—autoantibodies predict characteristic, immunotherapy-responsive syndromes. Pathogenic antibodies (shown in purple) bind to the surface-exposed domains of LGI1 and CASPR2. Also, some autoantibodies against LGI1 bind to the domain which docks with its receptors, a disintegrin and metalloproteases (especially ADAM22/23). Autoantibodies that bind extracellular protein domains are pathogenic, manifesting with characteristic clinical syndromes including faciobrachial dystonic seizures, limbic encephalitis and neuromyotonia. Both LGI1- and CASPR2-antibody mediated syndromes classically affect elderly men in their sixth decade, who improve markedly with immunotherapies. HLA-DRB1*07:01 is strongly linked to patients with LGI1 antibodies, and HLA-DRB1*11:01 to patients with CASPR2 antibodies. The non-pathogenic antibodies (shown in red) have been shown to bind the intracellular domain of VGKCs, and some bind the I125-α-dendrotoxin (α-DTX) used in the original VGKC radioimmunoassays. The targets of other double-negative antibodies are unknown, but likely to be other intracellular epitopes. Double-negative VGKC antibodies are found in a wide array of clinical syndromes, including epilepsies, dementias and primary psychiatric conditions, which span wide age ranges and lack either a clear immunotherapy response or a specific HLA association.
CASPR2, contactin-associated protein like-2; HLA, human leukocyte antigen; LGI1, leucine-rich glioma-inactivated 1; VGKC, voltage-gated potassium channel.
SPLITTING THE COMPLEX: IDENTIFICATION OF LGI1 AND CASPR2, AND CLINICAL ASSOCIATIONS
Biochemical interrogation of proteins complexed to VGKCs identified that leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein like-2 (CASPR2) were the actual targets of patient autoantibodies in the immunotherapy-responsive syndromes: limbic encephalitis, Morvan’s syndrome and neuromyotonia.8 9 Also, a few patients had contactin-2 antibodies.8 Crucially, the extracellular-exposed domains of these proteins were the direct antigenic epitopes. Table 1 describes the characteristics of these molecules, plus the LGI1 receptors ADAM22 and 23.
Molecular characteristics of LGI1, CASPR2, and associated proteins
LGI1 is a secreted neuronal protein, known to link pre- and post-synaptic terminals, akin to a molecular scaffold.19 Patients with LGI1 antibodies are typically aged over 60, with a 2:1 male to female predominance. LGI1 antibodies are prominent in patients with limbic encephalitis, found in many with Morvan’s syndrome, and in a few with neuromyotonia.8 9 23 In addition, around 50% of patients with LGI1-antibodies show a highly specific seizure semiology—termed faciobrachial dystonic seizures.24 Faciobrachial dystonic seizures typically begin before the onset of the cognitive impairment that characterises limbic encephalitis and are largely resistant to antiseizure medications but respond well to immunotherapies, often within a few days. They provide an excellent example of a distinctive phenotype with a robust response to immunotherapies whose characteristics translate to the other frequent focal seizure semiologies in patients with LGI1-antibodies.24 25
CASPR2 is a neurexin family protein with a large extracellular domain. Patients with CASPR2 autoantibodies are very often elderly males (Table 2). CASPR2 antibody–positive patients often have neuromyotonia or Morvan’s syndrome, and some have forms of limbic encephalitis as well as neuropathic pain syndromes. All typically respond to immunotherapy. Also, in patients with neuromyotonia and, especially, Morvan’s syndrome there is an association with tumours, typically thymomas.8 9 23 Importantly, LGI1 or CASPR2 specificities are rare outside of these syndromes. Taken together, LGI1 and CASPR2 antibodies have strong and specific clinical associations, and unequivocal clinical value in accurately detecting treatable syndromes.
FEATURES ASSOCIATED WITH DOUBLE-NEGATIVE VGKC ANTIBODIES
More recently, data from several groups have clarified the clinical relevance of double-negative VGKC antibodies, that is, VGKC antibodies without LGI1 or CASPR2 reactivities.26–28
Frequency and highly heterogeneous clinical features
In the largest such series, testing of ~100 000 samples yielded 3910 (~4%) samples with VGKC antibodies. Only 256 of these 3910 (6.5%) showed concomitant LGI1 or CASPR2 reactivities.29 Other studies report comparable rates of LGI1/CASPR2 antibodies amongst VGKC-antibody positive results: ~20% in adults30 and, in children, <5%:31 both observations are consistent with our unpublished experience. Hence, double-negative VGKC antibodies appear to account for ~85% of routine VGKC antibody requests. Is there any clinical utility in detecting these common antibodies? The data suggest not. First, double-negative VGKC-complex autoantibodies occur in ~5% of healthy controls, complicating their interpretation especially with widespread testing.2 6 26 28 Second, and in marked contrast to the well-defined clinical phenotypes associated with LGI1 and CASPR2 autoantibodies (Figure 1, Table 2), double-negative VGKC antibodies associate with a constellation of syndromes without age or gender predilection. These range from epilepsies, non-neuropathic pain, Alzheimer’s disease, peripheral neuropathy and headache, to primary psychiatric syndromes and leptomeningeal metastasis.26–28 32 Hence, it appears that double-negative VGKC antibodies have limited clinical specificity. Third, the VGKC antibody titre is not a clinically reliable measure: double-negative VGKC antibodies can occur at very high titres (eg 400–3000 pM). Therefore, the titre itself does not help in identifying LGI1 or CASPR2 specificity.26 27 29 30 33 Indeed, in clinical practice the concept of a ‘clinically relevant’ VGKC antibody titre has created many misdiagnoses, often where finding the antibody has overruled the clinical diagnosis.33–36
Comparisons of autoantibodies against LGI1, CASPR2, and the double-negative VGKC antibodies
Immunotherapy response
Patients with LGI1 and CASPR2 autoantibodies respond strikingly to immunotherapy, with improvements in 96%–100% of patients with LGI1 antibodies and 86%–100% with CASPR2 antibodies.24–26 29 Hence, detecting LGI1 and CASPR2 antibodies has clear therapeutic importance. In marked contrast, patients with double-negative VGKC antibodies respond poorly to immunotherapy, with response rates equal to those of placebo studies, or to rates observed from patients with negative VGKC test results.26 28 44
Serological observations: intracellular binding and non-pathogenicity
As our clinical suspicions increasingly indicated that double-negative antibodies had little clinical relevance, we predicted that studying their antigenic targets would offer clinically-relevant insights.28 First, we observed that the double-negative serum IgGs did not bind to any surface determinants expressed on live hippocampal neurons. Second, no double-negative IgGs bound to the extracellular domain of HEK293T cell surface expressed VGKCs. However, in this preparation, around one-third bound to intracellular aspects of the Kv1.1/1.2/1.6 subunits.8 28 Finally, a small proportion of the double-negative VGKC antibody sera directly bound the non-mammalian α-DTX employed in the radioimmunoassay. Taken together, these findings established that double-negative VGKC antibodies often bind targets that, in vivo in humans, are inaccessible or unavailable, thus mitigating their pathogenic potential.
By contrast, LGI1 or CASPR2 antibodies typically show robust cell-surface reactivity to the native, mammalian target (Figure 2) and strong data support their in vitro and in vivo functionality. For example, LGI1 antibodies can disrupt interactions between LGI1 and its receptors ADAM22/23, internalise the LGI1–ADAM complex and trigger neuronal hyperexcitability and memory deficits in vivo passive transfer models.20 21 25 42 There is also clear evidence supporting the pathogenicity of CASPR2 antibodies with rodent passive transfer reproducing human pain manifestations, and evidence that the antibodies can disrupt CASPR2 interactions with contactin-2.22 43
{kind=link}
{kind=link}
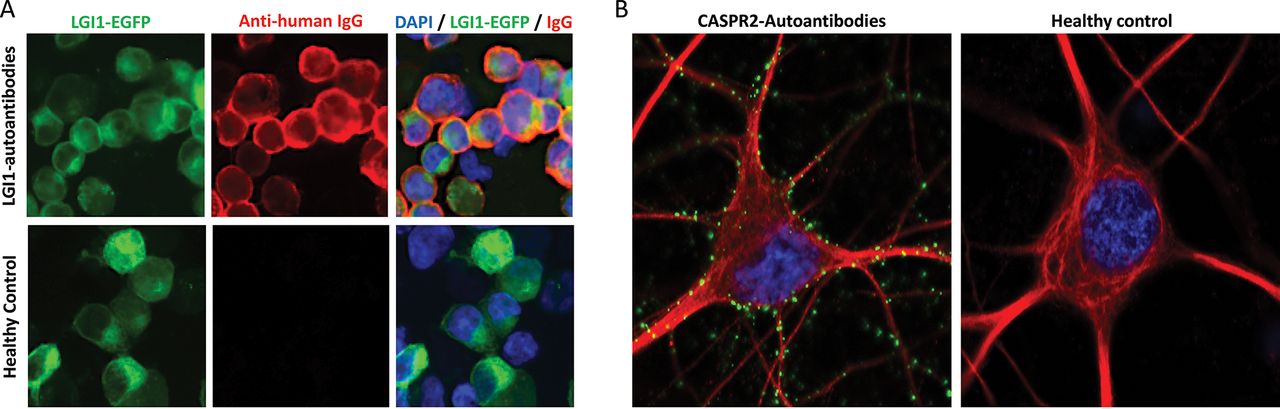
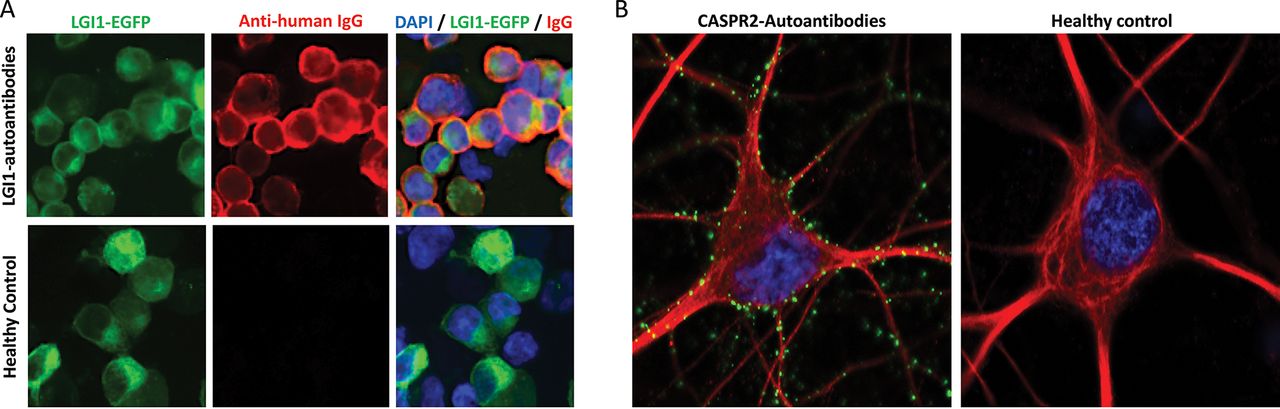
Cell surface-reactive autoantibodies measured by live cell-based assays or live hippocampal neuron binding. A. LGI1 antibody live cell-based assay. Serum from a patient with LGI1 antibodies binds to the surface of live LGI1-expressing cells (top row) but healthy control sera (bottom row) do not bind these cells. LGI1 tagged to enhanced green fluorescent protein (LGI1-EGFP, green) is expressed on the surface of live HEK293T cells; detected with anti-human IgG secondary autoantibodies (red); DAPI nuclear staining (blue). Images taken at ×40 magnification. B. Live hippocampal neuron assay. Human IgG autoantibodies (green) from a CASPR2 autoantibody-positive patient stain the surface of live hippocampal neurons (red, stained with an antibody against MAP2, microtubule-associated protein 2). Punctate green staining is observed along the dendrites and neuronal cell body. There is no neuronal reactivity when live hippocampal neurons are stained with healthy control serum (right). DAPI nuclear staining (blue). Images taken at ×63 magnification. In cases with seronegative syndromes, where the clinical picture is characteristic but the patient has negative results for the standard panel of known autoantibodies (LGI1, CASPR2, NMDA-R, GABAA/B-R, etc), live neuronal testing is an available diagnostic test which can be requested at our specialist laboratory. In this assay, any neuronal reactivity demonstrated by the serum/CSF, as above, strongly suggests the presence of a novel neuronal surface antibody and allows identification of novel antigenic targets.
CASPR2, contactin-associated protein like-2; CSF, cerebrospinal fluid; DAPI, 4′,6-diamidino-2-phenylindole; GABAA/BR, gamma-amino butyric acid A/B receptor; LGI1, leucine-rich glioma-inactivated 1; NMDA-R, N-methyl-D-aspartate receptor.
Genetic associations
Recently, a very strong immunogenetic basis was established for the diseases associated with LGI1 and CASPR2 antibodies.32 40 41 45 HLA-DRB1*07:01 and HLA-DRB1*11:01 were found to be over-represented in patients with LGI1 autoantibodies (~95% vs ~25% in healthy controls) and in those with CASPR2 autoantibodies (~50% vs ~10% in healthy controls), respectively. Extended haplotype associations were also observed.32 Moreover, the few patients with both LGI1 and CASPR2 antibodies carried yet another set of distinct HLA alleles.32 By contrast, there were no distinct HLA associations in patients with intracellular VGKC antibodies.32 Our recent clinical observations suggest the HLA analyses may form a useful adjunct to complement clinico-serological diagnoses (Irani and Waters, unpublished, 2020).
THE TEST MATTERS: LIVE CELL-BASED ASSAYS AND THE LIMITED SENSITIVITY OF VGKC ANTIBODY TESTING
Our understanding of test systems for detecting pathogenic antibodies has evolved. As only antibodies that can ‘see’ their targets in vivo have clear pathogenic potential, we prefer to use ‘live’ cell-based assays that present only extracellular domains of antibody targets. These avoid the detection of non-pathogenic species and the potential for contamination by fixative, which can alter target epitopes and permeabilise cells.46 Many diagnostic centres use fixed cell-based assays, largely for convenience. However, live testing remains biologically intuitive; it has a higher sensitivity and specificity, and can resolve samples that routine commercial kit testing assess as indeterminate.46–49 Such target-specific cell-based assays, where individual proteins are selectively expressed on the surface of live cells without detergent or fixative, are possible across the range of surface-directed autoantibodies, including LGI1 and CASPR2⇓ (Figure 2A).8 These live cell-based assays have identified up to 15% of samples from patients with immunotherapy-responsive illnesses that have detectable LGI1 or CASPR2 antibodies but no VGKC-complex antibodies.50 51 Hence, direct LGI1 and CASPR2 antibody testing should be performed as first-line testing, not as a second-line reflexive test after a positive VGKC antibody result.
SO, IS THERE ANY VALUE IN VGKC ANTIBODY TESTING?
The above evidence strongly argues against VGKC antibody testing in routine clinical practice. Yet, these antibodies may have some limited value in the research setting. First, cohorts with positive VGKC antibodies may have high rates of malignancy (12%–47%), suggesting VGKC antibody as an onconeural marker.33–36 However, such findings probably reflect an inherent clinical referral bias. Indeed, other reports suggest comparable tumour rates among patients with double-negative VGKC antibodies and matched VGKC-negative controls.26 Second, swine abattoir workers frequently have detectable, typically double negative, VGKC antibodies.39 While this is only rarely a vocational issue, it suggests that inhaled aerosolised brain tissue may be a model to study human autoimmunisation, although with resultant non-pathogenic reactivities. Interestingly, this observation suggests an inherent liability to a loss of immune tolerance against VGKC-complexed proteins may account for the high rates of VGKC antibodies occurring as secondary effects of multiple disparate disease processes.28 52 53 Finally, some observations suggest double-negative VGKC antibodies may co-exist with cell-surface autoantibodies.23 30 54 In this clinical context, the surface-directed antibody is likely to be directly pathogenic. Indeed, the high reported rates of VGKC antibodies in some diseases, such as neuromyotonia, may provide a lead for identification of samples with co-existent pathogenic reactivities.
CONCLUSIONS: A CALL TO CHANGE CLINICAL PRACTICE
In summary, double-negative VGKC antibodies usually target intracellular epitopes and lack pathogenic potential. They form the majority of the results in routine VGKC antibody testing. Importantly, and in stark contrast to finding antibodies directed against LGI1 and CASPR2, they do not predict a response to immunotherapy. Their detection in heterogeneous, often non-immune, clinical disorders at similar rates to healthy controls casts doubt on their detection in classic autoimmune scenarios. For example, this background rate is likely to the explain the common query that “my patient with Guillain–Barré syndrome / limbic encephalitis / Lambert–Eaton myasthenic syndrome has a double-negative VGKC of 1312 pM”. Further, the VGKC antibody radioimmunoassay is not an effective screening test for the key pathogenic autoantibody species as it misses ~15% of LGI1 or CASPR2 antibodies.
Taken together, the evidence from multiple international groups suggests there are no longer clinical reasons to test for VGKC antibodies. Stopping this test will reduce clinically irrelevant results, improve diagnostic accuracy and limit the use of unnecessary, potentially toxic, immunotherapies in patients. Most ‘non-immune’ syndromes referred to our specialist centre are associated with ‘double-negative’ VGKC antibody results. Yet, several laboratories continue to offer VGKC antibody testing, sometimes as a screening test to prompt LGI1 and CASPR2 antibody testing. Presenting local laboratories with the data and conclusions herein should encourage a change in testing regimens, towards patient benefit.
While this may be one of the final articles regarding VGKC antibody testing, work on LGI1 and CASPR2 antibodies will probably continue for several years, as the underlying immunology, autoantibody biology, and the molecular effector pathways still require clarification. These outputs could all impact on patient care by offering opportunities towards highly focussed examples of precision medicine. Nevertheless, for now, the evidence presented above should offer a marked improvement to the routine care of many patients each year.
Key points
Patients with pathogenic LGI1 or CASPR2 autoantibodies show distinct clinical syndromes and excellent responses to immunotherapy.
In contrast, those with non-pathogenic VGKC antibodies but with no LGI1 or CASPR2 reactivities have heterogeneous clinical syndromes and poor responses to therapy.
Testing for VGKC antibodies is deleterious for patient care and should cease.
By contrast, directly testing for LGI1 and CASPR2 antibodies can offer clear clinical benefit, with an accurate diagnosis and the chance of disease-modifying therapies.
Acknowledgments
SRI is supported by the Wellcome Trust (104079/Z/14/Z), The UCB–Oxford University Alliance, BMA Research Grants—Vera Down (2013) and Margaret Temple (2017), Epilepsy Research UK (P1201) and by the Fulbright UK–US commission (MS-Research Society Award). The research was funded/supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC; The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health). PW is supported by the UK NMO commissioning group. We thank Dr Antonio Berretta for his contribution to live neuronal staining images used within figure 2.
REFERENCES
Footnotes
Contributors SRI and SM drafted the original manuscript. All authors designed the figures, and reviewed and edited the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests SRI and PW are coinventors on ‘A Diagnostic Strategy to improve specificity of CASPR2 antibody detection.’ Ref. JA94536P. SRI is a co-applicant and receives royalties on patent application WO/2010/046716 (U.K. patent no. PCT/GB2009/051441) entitled ‘Neurological Autoimmune Disorders’. The patent has been licensed for the development of assays for LGI1 and other VGKC-complex antibodies. He has received honoraria from UCB, MedImmune, ADC Therapeutics and MedLink Neurology, and research funding from UCB, ONO and CSL Pharmaceuticals. PW is a named inventor on patents for antibody assays and has received royalties. He has received honoraria from Biogen Idec, Mereo BioPharma, Retrogenix and UBC; travel grants from the Guthy-Jackson Charitable Foundation.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed by Mike andi, London, UK.
Other content recommended for you
- LGI1, CASPR2 and related antibodies: a molecular evolution of the phenotypes
- Delayed LGI1 seropositivity in voltage-gated potassium channel (VGKC)-complex antibody limbic encephalitis
- Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies
- Emergence of new-onset psychotic disorder following recovery from LGI1 antibody-associated limbic encephalitis
- Occult bowel cancer presenting as Morvan syndrome
- Neuronal autoantibodies in mesial temporal lobe epilepsy with hippocampal sclerosis
- Anti-voltage-gated potassium channel complex antibody–mediated limbic encephalitis: a case report of a 53-year-old man admitted to intensive care psychiatric unit with psychotic mania
- LGI1 antibody encephalitis: acute treatment comparisons and outcome
- Voltage-gated potassium channel limbic encephalitis presenting as functional cognitive impairment
- Persistent anterograde amnesia following limbic encephalitis associated with antibodies to the voltage-gated potassium channel complex