Article Text

Abstract
New-onset refractory status epilepticus and its subcategory febrile infection-related epilepsy syndrome are rare devastating clinical presentations in those without pre-existing relevant history, often in schoolchildren or young adults, without a clear cause on initial investigations. A cause is later identified in up to half of adults, but in many fewer children. Patients often require protracted intensive care and are at significant risk of dying. Functional disability is common and subsequent chronic epilepsy is the norm, but some people do have good outcomes, even after prolonged status epilepticus. Patients need prompt investigations and treatment. Anaesthetic and antiseizure medications are supplemented by other treatment modalities, including the ketogenic diet. Despite limited evidence, it is appropriate to try to modify the presumed underlying pathogenesis with immune modulation early, with a more recent focus on using interleukin inhibitors. Optimising management will require concerted multicentre international efforts.
- epilepsy
Statistics from Altmetric.com
Introduction
Status epilepticus is a life-threatening emergency, especially if refractory.1 While immediate treatment of seizures is paramount, it is also important to establish the cause. Up to 20% of patients with refractory status epilepticus have a negative initial workup; these represent up to 60% of de novo refractory status epilepticus, and most are previously healthy young adults and children.2 Such cases have been variously referred to as having new-onset refractory status epilepticus (NORSE), idiopathic catastrophic epileptic encephalopathy or febrile infection-related epilepsy syndrome (FIRES). They have also been classified as ‘possible encephalitis’, since some have a preceding febrile illness and the diagnosis of autoimmune and infective encephalitis does not require an identified pathogen or antibody.3 4
Case history
After 3 days of a febrile illness, a previously well 19 years old developed focal seizures with loss of awareness. The seizures progressed to generalised tonic-clonic seizures and convulsive status epilepticus requiring sedation and ventilation in intensive care. Initial detailed investigations did not identify a cause and the status proved refractory despite antiseizure and anaesthetic medications, antiviral treatment and immune modulation. The patient died after 3 weeks of intensive care. The family were shocked and bereft: though informed of the gravity of the situation, they had not appreciated that this was a possible outcome.
Definitions
While earlier literature generally used NORSE for adult patients and FIRES for children, working definitions do not make this distinction, and both terms now apply in all ages. An international consensus meeting5 standardised the terminology for these disorders (Box 1). FIRES is now considered a subcategory of NORSE, irrespective of age. Presentations in children and young adults appear to share common pathophysiological mechanisms.5 6
Definitions
Consensus definition of new-onset refractory status epilepticus (NORSE)5
‘New-onset refractory status epilepticus is a clinical presentation, not a specific diagnosis, in a patient without active epilepsy or other pre-existing relevant neurological disorder, with new-onset of refractory status epilepticus without a clear acute or active structural, toxic or metabolic cause. This includes patients with viral or autoimmune causes. If no cause is found after extensive evaluation, this is considered “cryptogenic NORSE” or “NORSE of unknown cause”’.
Consensus definition of febrile infection-related epilepsy syndrome (FIRES)5
‘FIRES is a subcategory of NORSE that requires a prior febrile infection, with fever starting between 2 weeks and 24 hours prior to onset of refractory status epilepticus, with or without fever at onset of status epilepticus’.
Incidence, prognosis and mortality
NORSE is rare, typically occurring in previously healthy young adults and school-aged children. There are reports on approximately 200 NORSE and >200 FIRES cases.7 While there are no published figures on the incidence of NORSE/FIRES, the incidence of refractory status epilepticus is 3.4–7.2 per 100 000 per year8 9 and NORSE represents about 20%. In adult series there were more women, but among children there were more male. Older adults aged over 60 years occasionally develop the condition.
Published outcomes usually include a compound metric of functional outcome (eg, modified Rankin score), chronic epilepsy and cognitive impairment. Most NORSE cases become super-refractory. The use of anaesthesia, or the need to use anaesthetics, carries an increased risk of complications and poor outcome, especially with more anaesthetics used, concomitantly or sequentially, reflecting the severity of the status epilepticus.10 ,11 The median stay in intensive care for adults is 15 days12 and 20–40 days for children. Status epilepticus lasts longer in patients with cryptogenic NORSE, but with similar length of intensive care unit stay, outcome and mortality rates.12 Mortality in children is ~12%13 and in adults is 16%–27%.12 14 15 The functional outcome is usually poor, with half to two-thirds having cognitive and functional impairment (including vegetative state), and most having refractory epilepsy.12 14 16–19 Although a longer duration of status epilepticus and a higher score on the Status Epilepticus Severity Scale20 are associated with poorer outcome, patients can still have good functional outcomes and so prolonged treatment is justified. There are infrequent reported cases of a good outcome after months of refractory status epilepticus and coma.1 12 Nevertheless, the literature and our own experience show that it is insufficient to treat the status epilepticus alone without attempting to modify the underlying pathogenesis.
Clincal presentation, cause and hypotheses on pathophysiology
NORSE typically presents (in up to 90% of cryptogenic cases) in healthy people with a prodrome of a mild, non-specific influenza-like illness with headache, gastrointestinal or upper respiratory symptoms.12 21 Fever—required for the diagnosis of FIRES—precedes the illness in up to 60% of adults.12 Prodromal symptoms can precede seizures by 1–14 days12 13 sometimes with an asymptomatic interval in between. The most common seizures are ‘focal to bilateral tonic-clonic’ (secondary generalised).12 13 22 23 Brief and infrequent seizures build up over hours to days, sometimes reaching hundreds a day, evolving into super-refractory status epilepticus.
A cause is subsequently identified in up to half of adult patients with NORSE. Status epilepticus has some 200 uncommon causes,24 25 classified into inflammatory/autoimmune, unusual infections, genetic and metabolic/toxic. Autoimmune encephalitis, sporadic or paraneoplastic, is the most common cause identified in an adult.12 26 27 In children, there is no antibody consistently associated with NORSE/FIRES, despite occasional individual reports.28–32 However, some series predate the description of immune-mediated encephalitides. Antibodies against the N-methyl-D-aspartate (NMDA) receptor and the voltage-gated potassium channel complex are the most common in adult series, highlighting the importance of an autoimmune workup. Some autoimmune causes have characteristic clinical features. Patients with anti-NMDA-receptor encephalitis develop behavioural/psychiatric symptoms, movement disorder and seizures,33 34 associated in half of cases with ‘extreme delta brushes’ on electroencephalography (EEG).35 36 Encephalitides due to anti-LGI1 antibodies present as limbic encephalitis, sometimes preceded by facio-brachial dystonic seizures and inappropriate secretion of antidiuretic hormone.
Although cryptogenic NORSE has no distinguishing features, there are some findings that can help to distinguish it from anti-NMDA receptor encephalitis. One study found that patients with cryptogenic NORSE (n=11), when compared with those with anti-NMDA-receptor encephalitis (n=32), had more frequent prodromal fever, symmetric MR brain imaging abnormalities (including increased diffusion-weighted images or T2/fluid-attenuated inversion recovery (FLAIR) signals in the hippocampus, amygdala, insula, claustrum, thalamus, perisylvian operculum and basal ganglia in eight patients), less frequent involuntary movements, absent psycho-behavioural symptoms and absent cerebrospinal fluid (CSF) oligoclonal bands.21 The single episode of status epilepticus in NORSE can last weeks or months, distinguishing NORSE from explosive onset chronic epilepsy. Drug-refractory epilepsy commonly follows status epilepticus in NORSE without a latent period, unlike brain insults caused by herpetic and bacterial meningo-encephalitis or trauma, where epilepsy may also develop after a variable latent period, sometimes years.37 38
While fever-associated seizures develop in certain genetic conditions, such as Dravet syndrome, and with the metabolic stress of inborn errors of metabolism or mitochondrial disorders, seizures in cryptogenic NORSE/FIRES generally occur after rather than during the fever with a single severe episode of status epilepticus.39 Series testing in the UK and Europe of common epilepsy genes in cryptogenic NORSE and FIRES (6 adults and 15 children), including SCN1A, PCH19, POLG1 or whole exome sequencing, found no causative mutations.23 40 Small series in Japanese children reported mutations in SCN1A and SCN2A genes.41–43 While it is worth pursuing genetic causes of NORSE, ‘epilepsy genes’ are probably not a common cause.
Children with FIRES may have increased CSF concentrations of proseizure cytokines (eg, interleukin 6 (IL-6), tumour necrosis factor (TNF)-alpha) and chemokines44; adults with cryptogenic NORSE have increased concentrations in both CSF and serum.45 This supports the hypothesis that dysregulated immune hyperactivation may occur in those genetically predisposed in the aftermath of a trigger, possibly infective. A Japanese study found FIRES in children was associated with mutations in the IL-1 receptor antagonist gene (IL1RN).43 Release of cytokines and chemokines requires activation of T-cells, glia and perivascular cells, perhaps explaining the latent period between the prodrome and seizures.39 It remains unknown whether intrathecal inflammation is a cause or consequence of seizures. Only seven NORSE cases have been reported with postmortem neuropathology,14 46 47 and most have been unrevealing, showing only mild reactive changes (non-specific gliosis and microglial activation with patchy neuronal loss) without features of infection, vasculitis, infiltration or cortical dysplasia. Neuropathology in a young adult with fulminant presentation to our unit with negative virology showed a grey matter encephalitis, predominantly of T-cells.
Diagnosis and investigations
The recognition of NORSE/FIRES is reportedly typically delayed39 after an extensive search for underlying causes, delaying treatment. However, in our experience, these can be recognised early and application of the consensus definitions can encourage early recognition,5 preferably within the first 48 hours after status epilepticus onset. Several publications suggest a timed stepwise approach to investigations and management.7 48 Table 1 with a diagnostic work-up and the management flowchart (figure 1) are from the NORSE Institute website updated in 2020 (http://www.norseinstitute.org/definitions).
Investigations/diagnoses to be considered

Flow: adapted with permission from the NORSE Institute (http://www.norseinstitute.org/). The chart is from the last update of September 2020.
Early investigations (blood tests, brain imaging, CSF analysis and EEG), within 24–48 hours, can rapidly exclude important and treatable or reversible structural, infective, toxic and metabolic causes. Second-stage investigations may identify rarer causes. To date, there is no serological or CSF marker, only a non-specific mild CSF pleocytosis <10 cells/µL (≤5) and a slightly raised protein in up to two-thirds of patients. MR scan of brain and EEG can show suggestive features.39
MR brain imaging
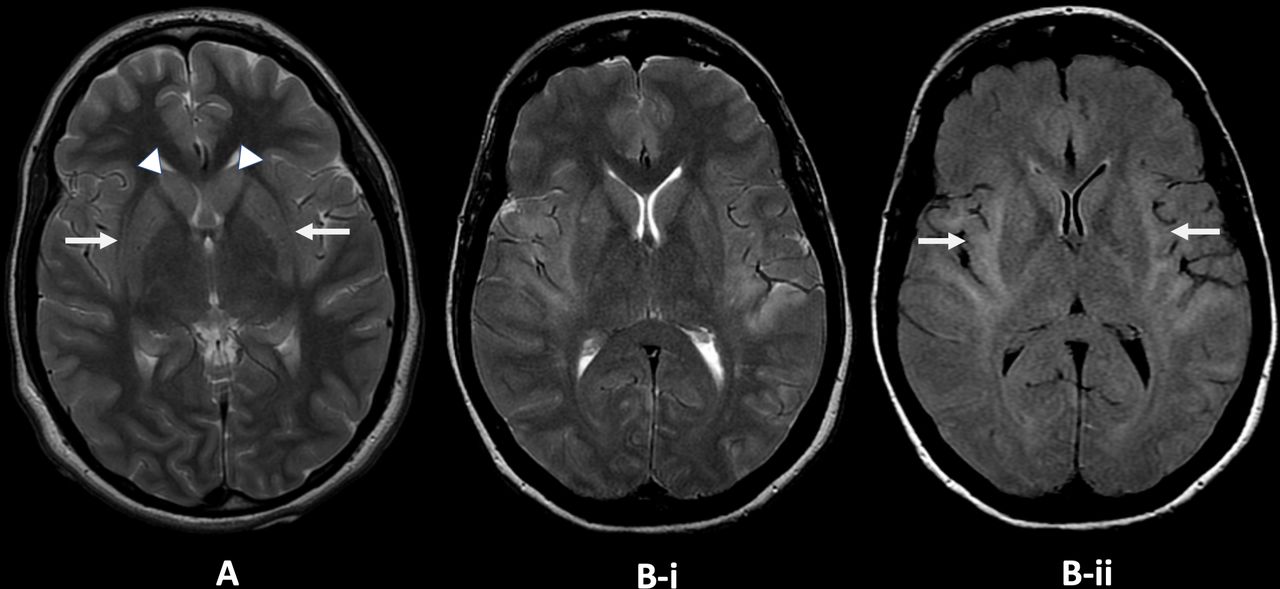
Non-specific abnormalities on MR brain scan include most commonly (on T2/FLAIR sequences) hyperintensity in limbic and medial temporal or neocortical areas, sometimes bilateral,12 with signal changes in the basal ganglia or the peri-insular region.37 49–52 Repeat imaging later may show hippocampal or generalised atrophy.22 23 49 51 In six children with cryptogenic FIRES,27 50 MR scan of brain was normal at disease onset (days 0–2 of status epilepticus), but soon (days 3–10) showed characteristic claustrum high signal (‘claustrum sign’) on T2/FLAIR and diffusion-weighted imaging, with normal apparent diffusion coefficient without contrast enhancement. This resolved after the status epilepticus subsided. None of the other 105 children with refractory status epilepticus had these changes. There have been other cases with similar early claustrum changes, including in adults (figure 2, and reviewed in Meletti et al 27). The claustral changes, however, were reported as infrequent, occurring in 3/39 cases in a recent series.51 MR abnormalities were observed in 82%, including mesial temporal lobe signal abnormality, diffuse leptomeningeal enhancement and later hippocampal and diffuse cortical atrophy. Leptomeningeal enhancement was associated with poor functional outcome and pharmacoresistant epilepsy, as were hippocampal and cortical atrophy, the former related to the duration of status epilepticus.
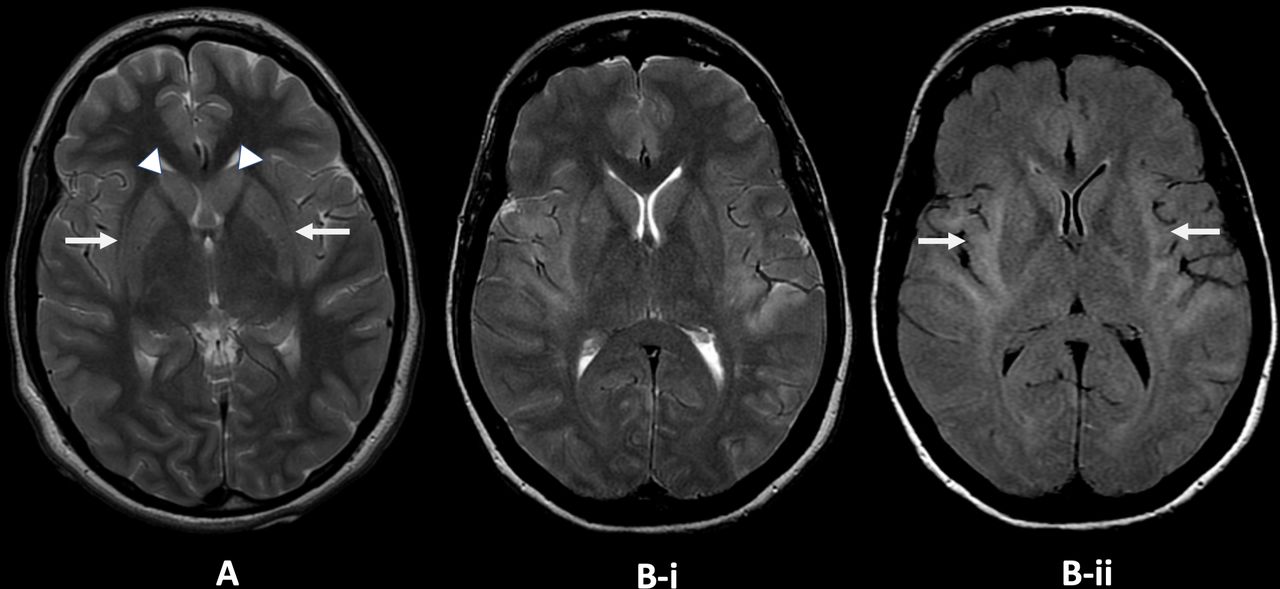
Claustrum changes on MR in two adult patients from the King’s College Hospital new-onset refractory status epilepticus database. (A) Axial T2-weighted image, patient 1: there is faint T2 high signal in the claustrum on both sides (arrows) with faint T2 high signal and slight swelling in both caudate nuclei (arrowheads). (B) Axial T2-weighted (i) and fluid-attenuated inversion recovery (ii) images, patient 2. There is T2 high signal involving the claustrum, external capsule and insula and temporal opercula on both sides (arrows). Image courtesy Dr Jo Jarosz, King’s College Hospital.
Electroencephalography
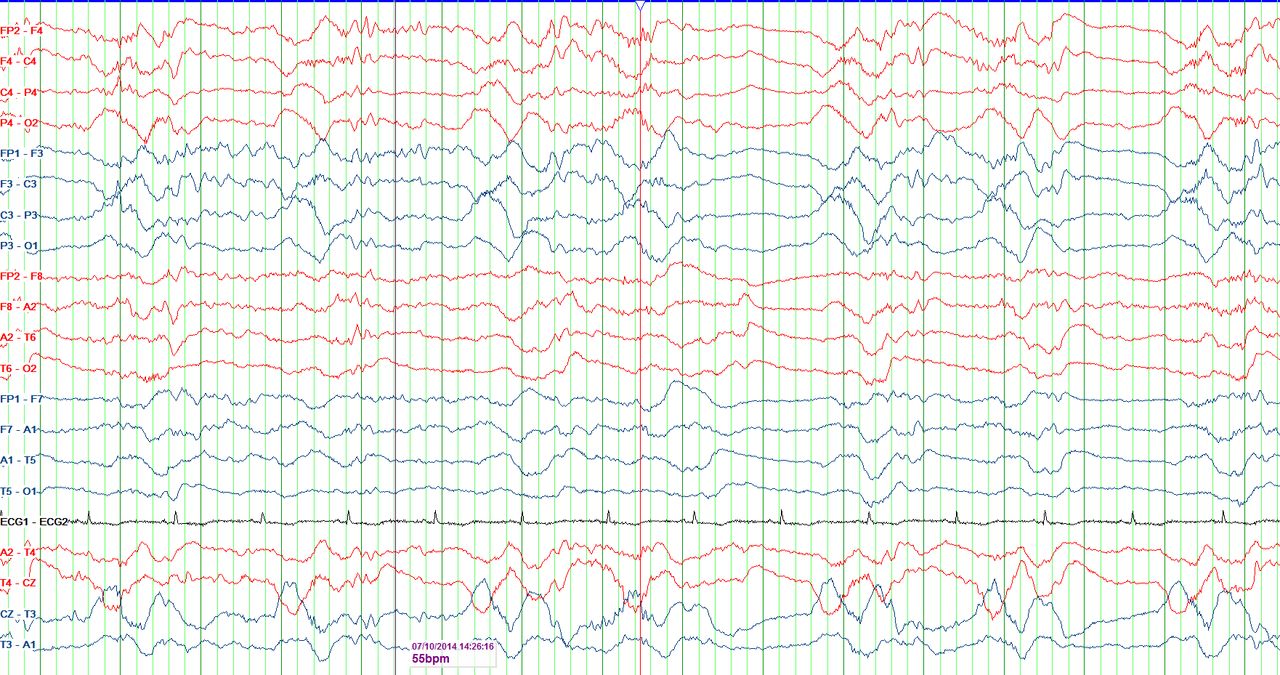
EEG is essential to monitor disease evolution and guide treatment. With so few studies of hyperacute EEG changes, there are no specific diagnostic or prognostic EEG markers. A retrospective study in children with FIRES identified three common early patterns: initially infrequent seizures gradually evolving into status epilepticus; a beta-delta complex resembling delta brushes (figure 3); a characteristic seizure onset pattern with focal fast activity with shifting seizures.53 There is no comparable study in adults. Most series reported periodic discharges (generalised, lateralised, bilateral independent and multifocal) and multiple seizure patterns (generalised, focal and multifocal).12 23 We have seen generalised patterns with focal onsets, as well as later evolution from generalised discharges to more focal or multifocal discharges/seizures as the condition settles. We have also frequently seen stimulus-induced rhythmic, periodic or ictal discharges (SIRPIDS), as reported in similar intensive care settings54 as well as in NORSE.55
{kind=link}
{kind=link}
{kind=link}
Electroencephalography (EEG): a periodic pattern can be seen consisting of delta waves preceded by fast activity and followed by electrodecrement in this routine EEG (bipolar montage) performed on an adult female patient with new-onset refractory status epilepticus. Abeta-delta complex resembling extreme delta brushes is highlighted (red line). Image courtesy Marisa Pinada Pinto, King’s College Hospital.
Brain biopsy
Brain biopsy to aid diagnosis is rarely reported, with little evidence to support its routine use. A series of 22 children with FIRES reported seven brain biopsies showing gliosis but no inflammation.56 One patient with NORSE triggered by primary angiitis of the nervous system was diagnosed on brain biopsy,57 although an MR brain scan had shown suggestive multifocal, scattered small deep white matter lesions on FLAIR. In a case report of FIRES, pathology of the previously resected occipital lobe, the seizure focus, showed neutrophilic leucocytes, T cells and microglial infiltration with severe spongiosis.58 In 43 patients reported with refractory SE and encephalitis, three underwent brain biopsy, two showing only mild astrogliosis. The third identified meningoencephalitis with marked lymphocyte infiltrates in the cortex and overlying leptomeninges, together with microglial nodules.
Management
General considerations
Super-refractory status epilepticus is best managed in a neuroscience centre with access to diagnostic tools and appropriate facilities, including continuous (or frequent) EEG monitoring, neuroimmunology input or extracorporeal membrane oxygenation. Early recognition should allow prompt transfer, where possible. Management is challenging and requires multidisciplinary care. Status epilepticus can be protracted and difficult to control, and prolonged intensive care unit stays carry inherent risks. Anaesthetic agents require experience to maximise benefit and minimise harm. There is only limited evidence to guide management and an individual’s outcome is difficult to predict, ranging from excellent recovery to a low responsive state or death. The time course often necessitates involving rotating clinicians, though continuity of care can be maintained by ensuring a core group is involved throughout.
Controlling status epilepticus goes hand in hand with attempting to modify the presumed disease process, even where the cause is unknown. Antiviral agents are usually given to cover possible viral encephalitis, and vitamin supplementation with thiamine and pyridoxine are considered. Treating epileptic activity alone is insufficient and, despite limited evidence, it is important to attempt to suppress the disease process early.
Management of NORSE/FIRES includes:
Investigating for an underlying cause.
Controlling status epilepticus, while avoiding iatrogenic complications
Treating the presumed inflammatory/ immune mediated process
Supporting the patient’s family
Rehabilitation
Suppressing clinical and subclinical epileptic seizures
Seizures are managed with antiseizure medications, anaesthetic agents, ketogenic diet or neuromodulation, while supporting the patient and minimising systemic complications, especially as status epilepticus is often protracted.
Anti-seizure medications
At presentation, standard management of status epilepticus applies according to local protocol. Trial evidence is limited to guide choice of antiseizure or anaesthetic drugs, beyond benzodiazepines59–62 and the recent Established Status Epilepticus Treatment Trial (ESETT) showing equal benefit as second-line treatment from levetiracetam, phenytoin and valproate for established status epilepticus.63 Valproate must be avoided if certain mitochondrial disorders are suspected. Other antiseizure medications available intravenously include lacosamide,64 phenobarbital and brivaracetam.65 Enteral antiseizure medications may also have a role, including other sodium channel blockers, topiramate,66 perampanel,67 stiripentol,68 pregabalin and vigabatrin (www.norseinstitute.org) as well as cannabidiol.69 70 In ventilated patients, antiseizure medications that are not clearly beneficial are weaned aiming to limit the number of agents.
Anaesthetic agents
Many patients require multiple anaesthetic agents to achieve control.12 23 Status epilepticus often resumes on weaning anaesthetics.19 23 Anaesthetic agents commonly used include midazolam infusion (often at high doses, with tolerance occurring), propofol, thiopentone (pentobarbital in North America) and ketamine. Although the propofol infusion syndrome is well recognised, it is our experience that thiopentone given for more than limited periods carries significant risk of complications including hypotension, acidosis, changes in serum potassium concentration, infections and ileus, although it is difficult to attribute complications to a single agent in a complex situation. Ketamine in moderate doses has fewer adverse haemodynamic effects. Side effects need to be anticipated, doses adjusted, and agents cycled or withdrawn. Enteral clonazepam or another benzodiazepine, such as clobazam, may facilitate later weaning of high dose midazolam infusions. EEG monitoring is essential in assessing progress and guiding treatment. Where there is doubt about EEG interpretation, depth of sedation is lightened to allow clinical assessment.
It is important to emphasise the need for caution and maximal care to ensure that suppressing epileptic activity does not come at the expense of systemic stability. Importantly, the following treatment approaches should be considered early in the course of disease.
Ketogenic diet
Ketogenic diet has reported benefit in this situation. Paediatric case series highlight its dramatic efficacy in FIRES within a few days of achieving ketosis.17 71 72 It is high-fat, low-carbohydrate diet that mimics fasting and exerts both antiseizure activity (possibly through decanoic acid inhibiting excitatory AMPA receptors73) and anti-inflammatory action (reduced proinflammatory cytokine levels) and improves mitochondrial function.74 Case reports also show benefits in adults.39
Introducing the ketogenic diet successfully requires minimising carbohydrate content in drugs or other infusions (reviewed in Williams and Cervenka74). Factors hampering its introduction include propofol within 24 hours—contraindicated due to the risk of fatal propofol infusion syndrome75—pre-existing metabolic and haemodynamic instability, liver failure and not absorbing enteral feeds. The ketogenic diet is contraindicated in rare metabolic diseases. Screening for such disorders (urinary organic acids and acyl carnitine profile) should be requested, in anticipation, soon after admission (Box 2). If there is no improvement after 2 weeks of ketosis, the diet is considered unsuccessful and stopped.74 The diet should be considered early before gastrointestinal difficulties preclude it. Intravenous ketogenic diet has been used particularly in children, using available guidelines.76 Close monitoring of serum lipids and liver function are essential (Box 2).
Screening investigations before using the ketogenic diet
Serum: full blood count, clotting screen, renal and liver profile, calcium, phosphate, vitamin D, magnesium, glucose, lipid, acylcarnitine, thyroid function.
Urine: urine organic acids.
Also recommended: vitamins A and E, zinc, selenium, copper, vitamin B12, folate, ferritin.
Neuromodulation for NORSE/FIRES
Based on a few published case reports,77–79 desynchronising networks by deep-brain stimulation may improve seizure control. Two children with FIRES refractory to ketogenic diet, cannabidiol and first-line immunomodulation were treated with centromedian nucleus deep-brain stimulation,75 implanted on days 27 and 37; secondary generalised seizures ceased but recurred on interrupting deep-brain stimulation, although focal seizures continued. One patient improved on subsequently starting anakinra (a recombinant IL-1 receptor antagonist) and obtained good functional outcome, while the second had poor outcome.
Vagus nerve stimulation is increasingly used for refractory and super-refractory status epilepticus80 81 including one patient with NORSE.81 The stimulator was implanted on day 5 in a 25-year-old man, with cessation of status for 72 hours. Seizures recurred thereafter and the patient succumbed with medical comorbidities. Vagus nerve stimulation clearly helped in one case of NORSE in our unit when implanted 37 days into the illness for frequent focal seizures, after generalised status epilepticus had come under control.
Other treatment approaches
Therapeutic hypothermia is used for neuroprotective and anti-inflammatory effects after perinatal asphyxia and adult cardiac arrest. Mechanisms of action may include the reduction of proinflammatory cytokines and protecting the integrity of the blood–brain barrier.39 There is very little evidence relating to NORSE. Two children with FIRES were successfully treated with moderate hypothermia to 33°C in combination with immunomodulatory treatment.82 On current evidence this is not generally advocated in this setting.
A few reported cases received electroconvulsive therapy in super-refractory status epilepticus83–85 but it is difficult to draw conclusions.
Immunomodulatory treatment
The hypothesis of an immune/inflammatory pathogenesis in cryptogenic NORSE and FIRES prompted the use of immunomodulatory drugs. First-line medications typically include intravenous corticosteroid, intravenous immunoglobulins and plasma exchange. Second-line medications include cyclophosphamide, rituximab, tocilizumab (IL-6 inhibitor) and anakinra (IL-1 inhibitor), with more recent focus on the interleukin inhibitors. Other antiseizure interventions also exert anti-inflammatory action, including ketogenic diet and cannabidiol. There is no randomised controlled evidence to inform doses, order or timing of immunomodulatory treatment. There are over 220 reported cases of cryptogenic FIRES and 100 of cryptogenic NORSE treated with immunosuppression.12–15 23 37 First-line agents are disappointing, even if given early.39 Second-line agents are difficult to evaluate, often used later and combined with other treatments, mostly in case reports or small case series. There is a growing consensus, however, that these patients need to be treated early with potentially disease-modifying interventions.
On current limited evidence, in cases of cryptogenic NORSE/FIRES where the status epilepticus is continuing, our first-line immunomodulatory treatment is a course of intravenous methylprednisolone, plasma exchange then intravenous immunoglobulin. Where possible, this is followed by the ketogenic diet, and if there is no contraindication, an interleukin inhibitor. We would also consider rituximab if anything points to an autoimmune cause (see NORSE Institute flowchart addressing treatment options).
Although not specifically referring to NORSE, a systemic review of variables in autoimmune encephalitis found that delay in immunotherapy contributed to worse outcomes.86
Below, we outline published evidence on more commonly used second-line treatments.
Immunosuppressive treatment
Rituximab
Rituximab is a chimeric monoclonal antibody against CD20 expressed on the surface of B cells. It reduces CD19+ B-cells within one week but does not influence antibody-producing plasma cells.87 Small case series included a small number of patients with cryptogenic NORSE responding to intravenous rituximab.26 84
Cyclophosphamide
Cyclophosphamide is a powerful alkylating immunosuppressant agent. One retrospective case series of 11 cryptogenic NORSE patients reported good outcome (modified Rankin score score 0–2) in 4/5 patients treated with intravenous cyclophosphamide on days 20–59 after disease onset. The patient with poor outcome started this treatment late (day 173). Long-term outcome at last follow-up, however, was poor in 8/11 cases.21
Other immunosuppressants
Single cases of FIRES have received azathioprine or tacrolimus.39
Modifying the inflammatory cascade: interleukin inhibitors
Although increasingly available and used in some units, the role of cytokine measurements in guiding treatment remains unclear.
Anakinra
Anakinra, a recombinant analogue of the human endogenous antagonist of IL-1 receptor type 1, inhibits IL-1β, a proinflammatory cytokine involved in autoinflammatory disease with powerful proseizure properties in animal models.88 Anakinra shows antiseizure properties in epilepsy models.89 It is given subcutaneously, and is contraindicated in people with neutropenia, a common side effect. There are reports of four children with FIRES treated with anakinra77 90 91: treatment was started between 6 days90 and 18 months after disease onset.91 Two of the four children responded well, although one also received deep-brain stimulation. Anakinra reduced the seizure burden both when given in the early (from day 6) and late chronic phases (after day 54) up to 1.5 years after disease onset. Successful treatment with Anakinra was also reported in a 21-year-old with FIRES, on day 32 of admission, with seizure cessation within 24 hours.92
Tocilizumab
Tocilizumab is a humanised monoclonal antibody against the IL-6 receptor. It is used in rheumatoid arthritis, giant cell vasculitis, autoimmune encephalitis and in cytokine-release syndrome45 and more recently severe COVID-19. IL-6 mediates the acute phase response, activates B-cells and T-cells, mediates cytokine expression as well as transition from innate to acquired immunity. The role of IL-6 in ictogenesis is unclear. Epilepsy models show both proseizure and antiseizure effects, and seizures may increase IL-6 concentrations.93 A retrospective series45 of tocilizumab given intravenously over 2 weeks in seven NORSE patients refractory to first and second line immunotherapy with rituximab after a median of 25 days found a response in all but one, with resolution of status epilepticus within 2–10 days. Five developed significant side effects, including leucopenia and severe infection. Three patients had a good outcome with a trend towards better outcome in those treated early. Tocilizumab was also used in seven with NORSE, five with prior rituximab, with resolution of status epilepticus after one or two doses in six (median of 3 days). Two patients experienced severe infection.94 Tocilizumab does not cross the blood–brain barrier, but prolonged seizures may disrupt this.
Family support
Relatives face a distressing time with their loved one, often young and previously well, suffering a devastating illness with a very uncertain outlook. Information, support and regular updates are essential, provided with honesty and compassion, ideally delivered by a core group of clinicians, ensuring continuity. Possible outcomes need to be imparted (death, significant deficit or reasonable recovery, usually with epilepsy) as well as the often protracted nature of the illness. We share the limitations of current evidence without transferring the burden of responsibility for management decisions. Families need support well beyond the intensive care stay. Relatives should be offered meetings if the patient dies. Communication challenges are the focus of a 2019 publication, where the authors also suggest palliative care services involvement alongside ongoing therapies with curative intent.95
Outlook
NORSE and its subcategory FIRES are rare challenging presentations. Many factors may temper, or delay starting treatment, including regulatory and funding hurdles. There is difficulty in assessing responses to interventions, which patients often receive simultaneously or in close succession. Whilst reports suggest that immunomodulation should start early, there is no high-quality published evidence to support whether to target the innate/adaptive or the humoural immune response or both, or in what order to administer these or other strategies. Addressing uncertainties regarding optimal management, whether specific treatments or intensive care, requires concerted efforts by multidisciplinary teams and multiple centres.
There are many current efforts to establish multicentre FIRES and NORSE registries, including the SENSE registry (Austria, Germany, Switzerland),96 97 the North American Critical Care EEG Monitoring Research Consortium (acns.org) and the Pediatric Status Epilepticus Research Group (pserg.org). The NORSE Institute has an ongoing prospective observational study and a family registry, open to anywhere in the world. In the UK, we have brought together a NORSE-UK group in 2019 as a multicentre multidisciplinary network of clinicians treating adult patients. With no multicentre randomised controlled studies, agreed treatment protocols can be based on most recent evidence with outcomes audited regularly. Such a network will also facilitate a registry/analysis of NORSE/FIRES cases, develop resources for family support and provide a platform for multicentre case-based discussions. Working together, we hope to share knowledge and management strategies, support patients and their families, and move closer to an understanding of and cure for this devastating presentation.
Further reading
NORSE Institute (www.norseinstitute.org).
Gaspard N, Hirsch LJ, Sculier C, Loddenkemper T, van Baalen A, Lancrenon J, et al. New-onset refractory status epilepticus (NORSE) and febrile infection-related epilepsy syndrome (FIRES): State of the art and perspectives. Epilepsia. 2018 Apr;59:745–752.
Hirsch LJ, Gaspard N, van Baalen A, Nabbout R, Demeret S, Loddenkemper T, et al. Proposed consensus definitions for new-onset refractory status epilepticus (NORSE), febrile infection-related epilepsy syndrome (FIRES), and related conditions. Epilepsia. 2018 Apr;59:739–744.
Key messages
Investigate patients with new-onset refractory status epilepticus early to identify an underlying cause, if possible.
Inform the family of the gravity of the situation and potential outcomes.
Continue to treat actively even if status epilepticus proves protracted: some cases have a good outcome despite prolonged status of up to several months.
Focus on avoiding iatrogenic complications of anaesthetic agents; the aim is to suppress epileptic activity but not at the expense of systemic complications.
Rationalise antiseizure non-anaesthetic medications in ventilated patients on anaesthetic agents, withdrawing those not seen to be effective.
Screen for contraindications to the ketogenic diet early and start this promptly before it is no longer possible (eg, when gut complications ensue); consider carbohydrate intake with medications/infusions not just feeds.
Despite limited evidence, treat promptly the presumed underlying process with immune modulation. Link up with other centres to agree treatment protocols and audit outcomes regularly.
Identify an intensivist and neurologist to provide long-term continuity where status is protracted.
Relatives require regular information and support throughout the illness. If the patient dies, contact the family to provide support and address concerns.
Acknowledgments
The authors are grateful to Dr Jo Jarosz, Consultant Neuroradiologist and Marisa Pinto da Pina, Subspecialty Lead for Telemetry, King’s College Hospital for figures 1 and 2). The authors are also particularly grateful to Professor Lawrence Hirsch, Yale, for his helpful comments on the manuscript as well as permission to reproduce the flow chart and table 1 from the NORSE INSTITUTE Website.
References
Footnotes
Contributors LMR wrote the first draft and edited the manuscript. LN cowrote and edited the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned. Externally peer reviewed by Ley Sander, London, UK.
Linked Articles
- Editors’ commentary
Other content recommended for you
- Refractory status epilepticus arrested by vagus nerve stimulation
- New-onset refractory status epilepticus (NORSE) in paediatric patients: causes, characteristics and outcomes
- Inflammatory epilepsy (FIRES) and haemophagocytic lymphohistiocytosis (HLH): an adult case
- New onset refractory status epilepticus (NORSE) as the heralding manifestation of herpes simplex encephalitis
- Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management
- Refractory status epilepticus complicated by drug-induced involuntary movements
- Unusual cause of fever, vision loss and super refractory status epilepticus in association with simian virus 40 (SV40)
- Claustrum hyperintensity: a rare radiological correlate in Niemann-Pick disease
- Genotypes and phenotypes of DNM1 encephalopathy
- Autoimmune encephalitis: proposed recommendations for symptomatic and long-term management