Article Text

Abstract
Skeletal muscle channelopathies are a group of rare episodic genetic disorders comprising the periodic paralyses and the non-dystrophic myotonias. They may cause significant morbidity, limit vocational opportunities, be socially embarrassing, and sometimes are associated with sudden cardiac death. The diagnosis is often hampered by symptoms that patients may find difficult to describe, a normal examination in the absence of symptoms, and the need to interpret numerous tests that may be normal or abnormal. However, the symptoms respond very well to holistic management and pharmacological treatment, with great benefit to quality of life. Here, we review when to suspect a muscle channelopathy, how to investigate a possible case and the options for therapy once a diagnosis is made.
- cardiology
- channels
- clinical neurology
- EMG
- muscle disease
Statistics from Altmetric.com
Introduction
Skeletal muscle channelopathies comprise a group of rare genetic neuromuscular disorders caused by dysfunction of sarcolemmal ion channels that are critical for muscle membrane excitability.1 In simple terms, they affect the ability of skeletal muscles either to contract, causing muscle weakness, or to relax, causing myotonia. Skeletal muscle channelopathies are divided into the periodic paralyses and the non-dystrophic myotonias, based on whether the predominant clinical symptom is muscle weakness or myotonia. The hallmark of a skeletal muscle channelopathy is that symptoms occur in an episodic or paroxysmal fashion causing acute disability.2 As a group, they affect approximately 1 in 100 000 people in the UK.3
Myotonia (delayed muscle relaxation after contraction) is often described by patients as muscles ‘locking’, ‘sticking’ or ‘cramping’. Episodic weakness can vary significantly from one limb ‘not working or feeling right’ to a tetraparesis. Symptoms commonly affect the leg muscles and in the case of myotonia may be precipitated by sudden or initial movement, leading to falls and injury. Symptoms are also exacerbated by prolonged rest, especially after preceding physical activity, and changes in environmental temperature.4 Leg muscle myotonia can cause particular problems on public transport, with falls caused by the vehicle stopping abruptly or missing a destination through being unable to rise and exit quickly enough. These difficulties can limit independence, social activity, choice of employment (based on ability both to travel to the location and to perform certain tasks) and are often socially embarrassing. The relationship of symptoms with activity often leads to sedentary behaviour, in turn a risk factor for comorbidities for example, heart disease or stroke. Muscle channelopathies are also frequently painful, which together with impaired physical ability, significantly impacts on quality of life.5 6
There are readily available symptomatic treatments for muscle channelopathies that can usually successfully ameliorate the burden of morbidity.7 More often, the biggest barrier to improving a patient’s quality of life is a lack of diagnosis. However, the diagnosis is not always easy, the conditions are rare, and patients may be completely asymptomatic with an apparently normal initial physical examination. The diagnosis hinges on careful history taking arousing clinical suspicion, supported by an accumulation of examination findings (or lack of abnormality) and investigative clues. No single test can provide an answer in isolation.
Clinical history
Skeletal muscle channelopathies include the periodic paralyses and the non-dystrophic myotonias.
The periodic paralyses are hypokalaemic periodic paralysis, hyperkalaemic periodic paralysis and Andersen–Tawil syndrome.
The non-dystrophic myotonias are paramyotonia congenita, sodium channel myotonia and myotonia congenita.
These are genetic disorders, and figure 1 shows the causative genes and the resultant dysfunctional ion channels. In most cases, symptoms can be traced back to the first or second decade, highlighting the importance of taking an early life history. Sometimes, the symptom onset is delayed into the 30s or 40s and/or may only appear during certain life events, for example, pregnancy.

The skeletal muscle channelopathies—causative genes, dysfunctional ion channel and resultant clinical symptom.
The periodic paralyses
The names of hypokalaemic periodic paralysis and hyperkalaemic periodic paralysis relate to episodes of muscle weakness accompanying either high or low serum potassium concentrations.8 They can be further distinguished by the typical pattern and duration of attacks and symptom triggers that either drive up the serum potassium (foods high in potassium) or drive it down (carbohydrate meals)—see figure 2. Myotonia can also develop in hyperkalaemic periodic paralysis but the usual presenting complaint is episodic weakness.

Features that distinguish hypokalaemic and hyperkalaemic periodic paralysis.
When taking a history from someone with possible periodic paralysis, it is important to bear in mind several caveats. Despite being called periodic paralysis, not all weakness episodes are this dramatic. The most profound episodes do include tetraparesis and these cases are most likely to present to the emergency department with a deranged (often low) serum potassium. However, many patients describe more modest weakness, for example, they can stand but only furniture walk, or they can walk on the flat but feel weak and cannot run or climb stairs. Weakness often presents during a period of rest following physical activity. Patients commonly recall having played with friends during school lunch break, returning to sit at a desk for lessons, and then being unable to stand or walk. Sometimes weakness may seem to be task specific or affecting only one limb, for example, a weak dominant hand after a prolonged writing. In these cases, there is usually a history of more generalised weakness; however, these may be the only symptoms initially volunteered as they are the most troubling.
Hypokalaemic periodic paralysis characteristically presents in the early teens with episodes of weakness on waking and lasting several hours. This may result in being unable to attend school in the morning but appearing ‘normal’ by the afternoon, and so easily misinterpreted as school avoidance.9 Similar scenarios can lead to disciplinary procedures for working adults.
The recurring episodes of weakness in periodic paralysis are similar but not usually stereotyped. Stereotyped episodes would more suggest a paroxysmal movement disorder or episodic ataxia, in which patients may erroneously use the term ‘weakness’ when struggling to quantify a symptom that is difficult to describe, for example, dyskinesia.
Andersen-Tawil syndrome
Andersen-Tawil syndrome is the only skeletal muscle channelopathy to affect systems other than skeletal muscle—namely cardiac muscle and bony development. The classical syndrome is a triad of periodic paralysis (usually mirroring the hypokalaemic periodic paralysis variety), cardiac conduction defects and dysmorphic features.10 However, in many cases, the dysmorphic features are subtle and overlooked, and the skeletal or cardiac muscle symptoms may dominate the clinical picture obscuring the true breadth of systemic involvement. Patients affected predominantly by periodic paralysis may be misdiagnosed as having hypokalaemic periodic paralysis. This has important implications, as the patient’s cardiac risk and need for subsequent monitoring may be overlooked. Sudden cardiac death may occur in Andersen-Tawil syndrome but is generally considered rare. However, cardiac arrhythmias requiring medical intervention appear relatively common, with 20% of patients requiring an implantable cardiac defibrillator in one series,11 and sometimes occurring without prominent cardiac symptoms. We recommend all patients with Andersen-Tawil syndrome (regardless of symptoms) have annual cardiac review by an experienced cardiologist, with ECG and prolonged Holter monitoring.
The non-dystrophic myotonias
Myotonia is usually the presenting problem in both myotonia congenita and paramyotonia congenita. It can be difficult for patients to describe (especially before a diagnosis is made) and they may use terms such as muscles cramping, being stiff, stuck, aching, or ‘not working’. Myotonia can affect any skeletal muscle so although typical patterns of involvement occur—legs more than hands and face in myotonia congenita, hands and face more than legs in paramyotonia congenita—symptoms affecting the trunk, back (back pain) and chest muscles (cannot catch breath/take a deep breath) can all be problematic. Myotonia affecting the face can affect the jaw and tongue, making chewing or swallowing difficult especially with cold food or drink. Cardiac and smooth muscle (bowel or bladder) are not affected in the non-dystrophic myotonias.
Other characteristic pointers towards either myotonia congenita or paramyotonia congenita include the warm-up phenomenon in myotonia congenita (myotonia improving with repetitive activity) or paradoxical myotonia in paramyotonia congenita (worsens with repetition), as well as different forms of episodic weakness (see table 1). Sodium channel myotonia has features that overlap with both myotonia congenita and paramyotonia congenita but this term refers essentially to a pure myotonic disorder without episodic weakness, caused by mutations in the SCN4A (sodium channel) gene.
Clinical features of the non-dystrophic myotonias
Myotonia itself is a symptom and/or sign, not a diagnosis. It can be seen or detected during electromyography (EMG) in other conditions including myotonic dystrophy types 1 and 2, Pompe’s disease12 and other myopathies13 (where it may have no overt clinical signs and be detectable only by EMG), or it may be drug induced.14 Nevertheless, by far the most common diagnosis in someone with myotonia is myotonic dystrophy type 1; this should always be considered especially in younger patients who may not yet manifest its multisystem involvement. Also DM2, less common than DM1 and with more mild systemic features (even at older ages), can cause confusion with non-dystrophic myotonia.
Pain and fatigue
Although episodic weakness or myotonia are the dominant clinical symptoms in the skeletal muscle channelopathies, pain (myalgia) and fatigue are often prominent and sometimes are the patient’s main concern.5 6 15 It is important to enquire about these and to attempt to treat them as well as addressing weakness and myotonia.
Examination findings
The examination is typically normal in the periodic paralyses although some patients do develop a proximal myopathy. However, if patients are examined during an episode of weakness, as well as reduced strength, the reflexes are reduced or absent. In someone with hyperkalaemic periodic paralysis there may be detectable myotonia. Patients with Andersen-Tawil syndrome can have dysmorphic features including short stature, low-set ears, micrognathia, clinodactyly, dental abnormalities.16 Note that any dental abnormalities may have been corrected in childhood, so it is important to enquire about dental work, especially extraction for overcrowding.
People with myotonia often have generalised muscle hypertrophy, or well-defined muscles.9 Because they are often young adults at presentation, it is easy to assume they look this way because they are young and fit. Muscle hypertrophy must always be considered in context and clinicians should make a specific enquiry as to how active or how many times a week they actually attend the gym.
Patients with a myotonic disorder will (nearly always) have evidence of myotonia on examination but this can be easily overlooked in a general neurological examination. Patients with myotonia congenita are best diagnosed from the waiting room. They may well have sat for a while waiting to be called. When they first rise, their leg muscle myotonia causes them to be slow and to have a stiff gait. By the time they reach the clinic room this has become normal. Examination for eyelid closure or hand grip myotonia should be repeated. If the patient has paramyotonia congenita the first movements may appear unrestricted and only with repetition will myotonia become evident (paradoxical myotonia). Conversely in myotonia congenita, the myotonia may be immediately evident with the first test but improves or disappears with repetition that is, the warm-up phenomenon. We usually ask the patient to grip their hands tightly or to close their eyes ‘as if they have soap in them’ for 5 s, then to open the hands/eyes as fast as possible. This should be repeated three to five times. Other findings can include contractures, for example, at the elbow and scoliosis, although this is most commonly seen in children and young adults.
Investigations
Investigations performed when considering the possibility of a muscle channelopathy need to be interpreted in light of the overall presentation, as a negative test in isolation does not exclude a channelopathy.
Bloods tests/creatine kinase
The main role of blood tests in investigating a skeletal muscle channelopathy is to exclude other diagnoses. A standard ‘battery’ includes tests of renal function (is there renal disease altering the serum potassium?), thyroid function tests (does the patient have thyrotoxic periodic paralysis?), white cell enzymes (Pompe’s disease) and creatine kinase. Serum creatine kinase in people with skeletal muscle channelopathies typically ranges from being within normal limits to being up to 1000 IU/L.6 It is a non-specific test but an abnormal result always helps to support the suspicion of a neuromuscular disorder. However, a normal result does not exclude a skeletal muscle channelopathy. Values above 1000 IU/L can occur and are consistent with a skeletal muscle channelopathy diagnosis but they are less common17; values this high makes it worth considering the whole presentation and asking if this could reflect an alternative neuromuscular diagnosis (see table 2). There are rare reports of a skeletal muscle channelopathy with a serum creatine kinase above 10 000 IU/L and presentation with rhabdomyolysis.18
Differential diagnoses of skeletal muscle channelopathies
Ictal serum potassium concentrations can help but are not absolute in someone with possible periodic paralysis. If the weakness is severe enough to cause tetraparesis, then the serum potassium will usually be outside the normal range. However, the key element associated with weakness is a rise or fall in serum potassium concentration; the absolute value is not always abnormal. This can be especially true if an attack is beginning to subside, or has subsided by the time any blood tests are taken.
Neurophysiology
Standard EMG and nerve conduction studies are usually normal in the periodic paralyses (except in some cases of hyperkalaemic periodic paralysis with myotonia). The most informative test to diagnose a periodic paralysis is a long exercise test.19 This involves stimulating the ulnar nerve and recording a series of compound muscle action potentials (CMAP) from abductor digiti minimi before and after ‘exercise’ of the muscle (abduction against resistance over a 5 min period). The postexercise CMAPs are recorded for a 50 min period looking for a 40% or more decline in amplitude compared with the postexercise peak. A positive test indicates a periodic paralysis but not the subtype. In a significant minority of cases, especially in those with Andersen-Tawil syndrome as well as those with rare attacks of weakness, the long exercise test can be normal.20 So, a positive test can help but a negative test does not exclude a diagnosis of periodic paralysis.
EMG will determine the presence of myotonia. However, myotonia is non-specific and having identified it, the clinician needs to consider all the possible differential diagnoses (table 2). Electrical myotonia without clinical myotonia is more common in people with a myopathic disorder but usually there are other indicators of this, for example, a myopathic EMG, fixed weakness on examination and persistent functional disability. A repeat short exercise test can show certain patterns of response that help to indicate the different sub-types of non-dystrophic myotonia21 but it is not specific and there is overlap with the patterns seen in myotonic dystrophy. Now that next-generation sequencing allows genes to be sequenced in parallel, rather than having to select a ‘first choice’, this test has less practical value.
Genetic testing
Next-generation sequencing means that parallel testing of all genes relevant to skeletal muscle channelopathies is now available in the UK. This is particularly helpful for Andersen-Tawil syndrome, which may present clinically with a hypokalaemic periodic paralysis picture but an accurate diagnosis can be made only by next-generation sequencing. This includes analysis of all three periodic paralysis genes, SCN4A, CACNA1S and KCNJ2. Due to the cardiac risks of Andersen-Tawil syndrome we recommend offering genetic testing to all relatives of a proband, regardless of symptoms.
For a possible myotonic disorder, CLCN1 and SCN4A are also analysed in parallel. DM1 and DM2 genetic testing needs to be requested separately and should not be forgotten even if the patient appears ‘only’ to have a myotonic disorder.
In terms of inheritance pattern, all skeletal muscle channelopathies are autosomal dominant or de novo, except for myotonia congenita, which can be dominant or recessive. Recessive CLCN1 variants can cosegregate with an SCN4A variant or an expansion in the DMPK and CNBP genes associated with DM1 and DM2. This can cause diagnostic confusion, for example, if a proband has a clinical diagnosis of myotonia congenita but genetic analysis identifies one variant in a family with dominant inheritance of a myotonic disorder, it may be assumed that this reflects a positive diagnosis; in fact, the proband may have myotonic dystrophy but be a carrier for a recessive CLCN1 variant. This has clinical implications as the patient may not be monitored for cardiac and other systemic complications if diagnosed with myotonia congenita.
The characteristic phenotypes of the skeletal muscle channelopathies have been described for over 100 years and the genetic basis of these disorders known since the early 1990s. A significant minority of patients do present with a channelopathy like clinical presentation but have a negative genetic result for these established disease causing genes.
In the last few years, several alternative genetic causes of episodic neuromuscular weakness have been identified, with expanding phenotypes.22 23 Other neuromuscular conditions can also have episodic or fluctuant presentations, for example, metabolic disorders (see table 2); these should be considered if genetic testing for a skeletal muscle channelopathy is negative. Also other paroxysmal disorders, especially GLUT1 deficiency syndrome and paroxysmal kinesigenic dyskinesia, can erroneously be referred to a neuromuscular clinic.24
Other investigations—MR scanning and muscle biopsy
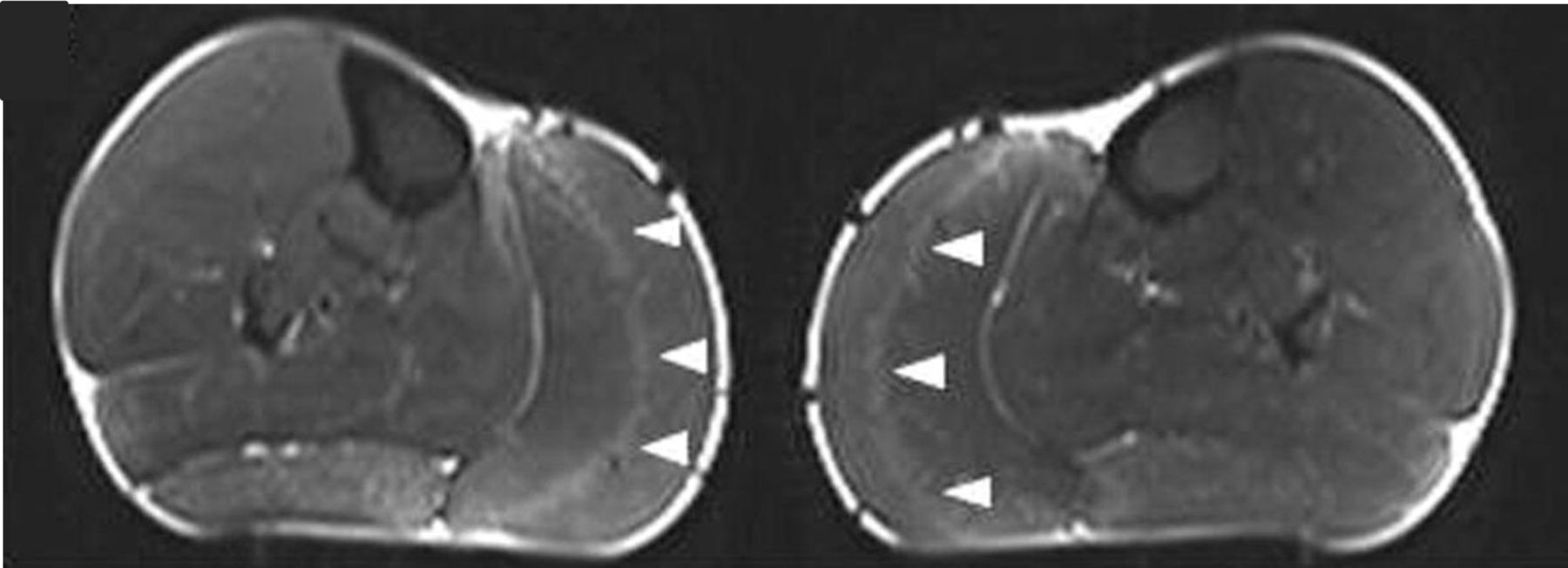
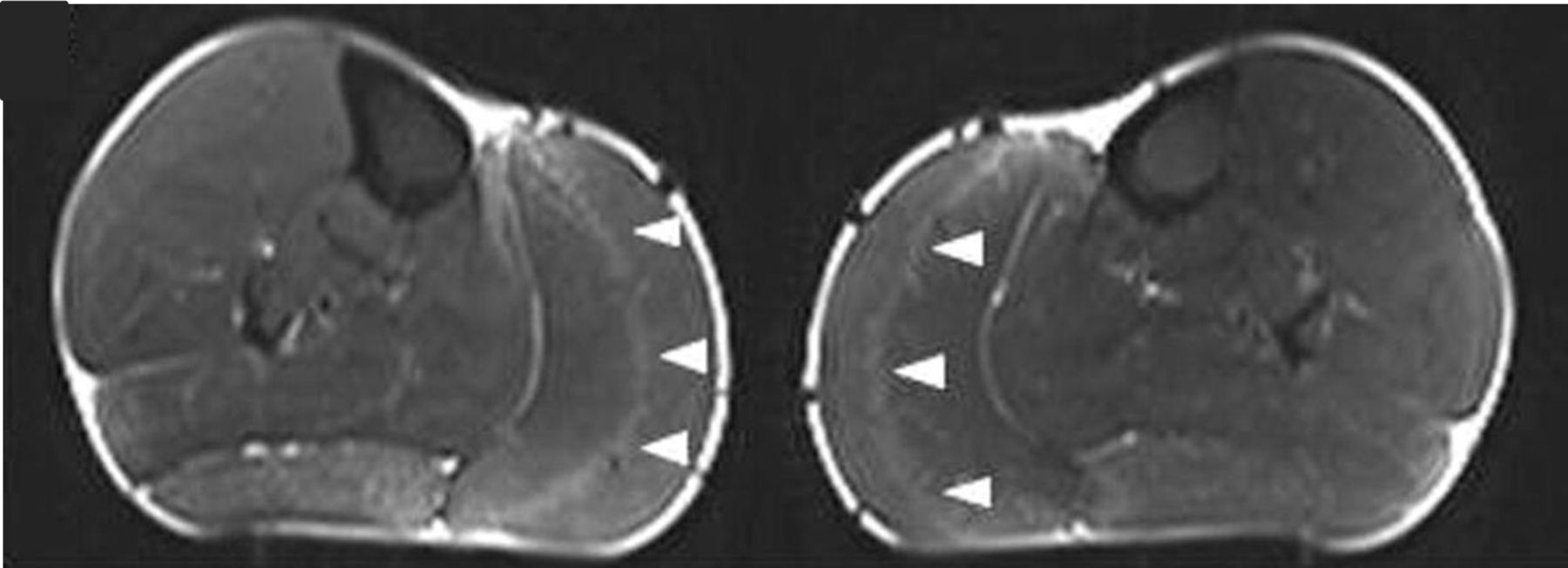
MR scanning of muscles, usually of the lower limb muscles in skeletal muscle channelopathies, can show fatty muscle infiltration in keeping with the clinical development of fixed myopathy25 or T1 STIR hyperintensity. In periodic paralysis the STIR hyperintensity probably reflects disease activity, that is, ongoing recurrent episodes of weakness. This can help in guiding symptom treatment and response. Early pattern recognition, including a gastrocnemius ‘central stripe’ in myotonia congenita26 may be diagnostic pointers (figure 3) but as yet there is no established definitive MRI pattern of muscle involvement for skeletal muscle channelopathies.
{kind=link}
{kind=link}
{kind=link}
MR scan images (STIR) of both calves in a patient with myotonia congenita, showing the ‘central stripe’ in both left and right medial gastrocnemius muscles (arrowheads). Figure reproduced.26
A muscle biopsy is not necessary or generally informative for the diagnosis of typical skeletal muscle channelopathies. Those performed historically have shown non-specific myopathic findings, including a vacuolar myopathy in periodic paralysis.27 Exceptions to this include the newer and atypical phenotypes of channelopathy, for example, RYR1-related periodic paralysis where a muscle biopsy can support the diagnosis.22
Management
Skeletal muscle channelopathies are genetic disorders that currently are not curable but there are numerous symptomatic treatments and management approaches that can significantly impact morbidity.
Lifestyle and activity
Many people with skeletal muscle channelopathies report some clear symptom triggers, for example, foods high in carbohydrate or potassium. Keeping a food diary can help to identify these and help people to avoid them or make alternative food choices.
The relationship between symptoms and exercise means that many people with these conditions avoid exercise and activity, having low levels of physical activity and difficulty maintaining an active lifestyle. However, those people who have managed to develop ways of staying active do find this helpful.
There are well-established benefits of keeping active, in terms of extending health span, and particularly for people with long-term conditions. There are also well known correlations for the general population between extended periods of inactivity (sedentary behaviour), health outcomes including obesity, cardiovascular and metabolic diseases, and an increased risk of mortality.28 29
There is little published evidence about activity specifically in people with skeletal muscle channelopathies, although our personal experience suggests a trend for high levels of sedentary behaviour in this population, indicating that interventions addressing this may help improve health outcomes.
Advice on extended periods of warm up and cool down, and stretches before and after exercise, can support an individual to transition in and out of, and prepare for exercise, without precipitating symptoms and/or in some cases associated muscle pain. Low intensity and low resistance exercises, focusing on gradually increasing repetitions and resistance as muscles adjust, can also help to avoid worsening myotonia or precipitating weakness. Activity monitors or apps can help to measure activity, and to establish a baseline from to set goals for gradually increasing activity and/or to interrupt extended periods of sedentary behaviour. Walking and moving around is often more accessible than formally working out at the gym.
Fatigue may also improve with these approaches or may require additional referral to a formal fatigue management programme.
Pharmacological treatments
Some people choose to manage symptoms by adjusting triggers as much as possible.7 When medication is indicated there are numerous choices. In the periodic paralyses, prophylactic treatment aims at controlling serum potassium, with diuretics that are either potassium sparing (aldosterone antagonists for example, spironolactone, or amiloride) or potassium wasting (thiazides). For those with hypokalaemic periodic paralysis, potassium supplements taken at the onset of an attack can help to abort or minimise symptoms.7 Other options effective in both forms of periodic paralysis include carbonic anhydrase inhibitors: acetazolamide or dichlorphenamide (not currently available in UK). Long-term acetazolamide requires an annual renal ultrasound scan to monitor for renal calculi. The treatment of non-dystrophic myotonia of any subtype is with sodium-channel blockers. Mexiletine is most widely used and has the greatest available evidence of efficacy and safety30–32 but lamotrigine is increasingly popular following a recent clinical trial.33 Other options with more limited or anecdotal data include ranolazine, carbamazepine, flecainide, propafenone and phenytoin. All options are aimed primarily at treating either episodic weakness or myotonia. There are no studies to indicate the best treatment of pain or myalgia in these conditions. Our experience is that acetazolamide, often combined with an anti-myotonic agent such as mexiletine can help in the non-dystrophic myotonias but that analgesics, including neuropathic agents, often have limited benefit.
There is some limited evidence that aggressive control of episodic symptoms might influence the longer term development of a proximal myopathy that can occur in muscle channelopathies.
Cardiac management
All patients with Andersen-Tawil syndrome should have an annual review with a cardiologist. Extended ECG recording, for example, 24 or 72 hours Holter monitoring may detect arrhythmias that require treatment even when the patient reports no symptoms. There is no randomised controlled trial evidence for the most effective antiarrhythmic regimen but beta-blockers or flecainide are commonly used. The requirement for interventional devices such as an implantable cardiac defibrillator appears to be more common than previously appreciated, emphasising the importance of expert cardiac involvement.11
Emergency treatment of periodic paralysis
Among patients with skeletal muscle channelopathies it is those with hypokalaemic periodic paralysis who present most commonly to emergency departments, due to the extended nature of their attacks. All patients presenting with a first episode of hypokalaemic periodic paralysis should be investigated for secondary causes of hypokalaemia, and especially thyrotoxicosis (figure 2). Those with a low serum potassium require ECG monitoring. Any ECG changes are an indication to use an intravenous rather than oral potassium replacement. ECG and serum potassium monitoring must continue during replacement and for several hours after its correction to normal. This is because during an attack, potassium is held intracellularly but the total body potassium is not actually low. As the attack subsides, potassium returns to the serum. Combining this with exogenous replacement can result in a rebound hyperkalaemia and iatrogenic death.34
Pregnancy and labour
Most pregnant women with skeletal muscle channelopathies report symptom exacerbation during pregnancy,35 36 probably due to both hormonal changes and medication withdrawal. The current recommendation is to stop medication before and during pregnancy due either to evidence of teratogenicity or a lack of safety data. There are some reports of mexiletine having been used without major adverse event during pregnancy37 38 and the more recent evidence of lamotrigine’s efficacy in myotonic disorders33 offers a potential option for pregnant women. Worsening symptoms can be particularly detrimental if they provoke increased falls. The symptoms may necessitate taking maternity leave early, with the knock-on effect of limiting time available to spend with baby post-partum.
Many women with skeletal muscle channelopathies have spontaneous unassisted vaginal deliveries, and this should remain an option when birth planning. However, we usually recommend considering the labour to be relatively high risk, such that delivery takes place in a facility where there are senior paediatric, obstetric and anaesthetic staff and facilities available if required. Symptoms of weakness or myotonia may prolong a labour and delivery with all the inherent complications of this, but also a subgroup of infants can have life-threatening respiratory complications if they have inherited the disorder39 (predominantly those inheriting an SCN4A gene mutation).
Anaesthetic considerations
For any patient requiring an anaesthetic (including during labour) it is essential the anaesthetist knows of the diagnosis of a skeletal muscle channelopathy beforehand, where feasible. Local or regional anaesthetics are preferred to a general anaesthetic if possible. If using general anaesthesia, propofol and non-depolarising anaesthetic agents appear to be effective and safe.40 Depolarising anaesthetics or suxamethonium can precipitate a myotonic crisis with profound muscle rigidity, which can mimic a malignant hyperthermia type reaction. If the rigidity includes the jaw muscles, intubation may be impossible.41 Suxamethonium can also induce hyperkalaemia and ventricular arrhythmia, which can be fatal.42 Emergency treatment for a myotonic crisis is with intravenous sodium channel blockers such as lidocaine.40
Note that other peri-operative factors can also exacerbate symptoms of a skeletal muscle channelopathy for example, prolonged immobility, or a cold operating theatre/recovery room.
In summary skeletal muscle channelopathies are rare. They can evade diagnosis because the symptoms are sometimes hard for a patient to describe, the examination is often normal and investigations may be normal or negative unless conducted during symptoms. Yet they are extremely treatable and making the appropriate diagnosis and management plan can be very rewarding for clinicians, and greatly benefit patients.
Case example: a weak patient in the emergency department with a low potassium—is it periodic paralysis?
History
The age of the patient can be indicative. Drug history may be more pertinent in older patients. Genetic disorders are more likely to present for the first time in younger patients, the typical age of onset of hypokalaemic periodic paralysis being in the teens. These are of course pointers only, and age is not an absolute criterion for genetic causes of periodic paralysis. Hypokalaemia from any systemic, drug-induced or genetic cause can induce muscle weakness. Ask the patient if they have diarrhoea and vomiting, tachycardia and palpitations (thyrotoxicosis). Is there a history of renal disease? Take a drug history and family history (is there a known family history of periodic paralysis or relatives with the same symptoms)? Has this happened before or have they had milder symptoms of weakness that resolved spontaneously without needing to attend the Emergency Department?
Examination
Weakness typically affects the limbs and trunk with reduced or absent reflexes. Muscles of the neck and face are less commonly affected but can be. This may cause slurred speech if the lower face and tongue are weak or for there to be eye closure weakness. Extraocular muscles are not usually weak; an inability to speak or to open the eyes at all is not typical and more suggestive of functional weakness. In young children with hypokalaemic periodic paralysis respiratory muscle weakness can cause respiratory distress and hypoxia. This is much less common in teenagers and adults but can occur in severe episodes with tetraparesis. Does the patient look dysmorphic? Do they have short stature? Short stature and dysmorphic features vary widely in Andersen-Tawil syndrome but among the most common are micrognathia (small chin), clinodactyly (curved digit, usually the little fingers) and low-set ears. Are there any other systemic abnormalities? Hypertension may accompany secondary causes of hypokalaemia. Tachycardia may suggest thyrotoxicosis but can occur with hypokalaemia itself, or may just indicate the patient is anxious about being unable to walk.
Initial investigations and management
Hypokalaemia with normal acid–base balance is expected in genetic forms of hypokalaemic periodic paralysis. Acid–base disturbance should prompt more extensive renal, and endocrine investigations for, for example, renal tubular acidosis, Gitelman’s syndrome, or hyperaldosteronism. Given that the hypokalaemic weakness may be the only indicator of thyroid disease we recommend routinely testing all first presentations. Serum creatine kinase can be normal or raised and is non-specific to diagnosis but a positive test can help with triaging an onward referral after the acute episode.
Monitor the ECG and serum potassium. There is no absolute value for when to choose oral vs intravenous replacement but a potassium <2.5 mmol/L, severe muscle paralysis especially if breathing is impaired, or ECG changes of hypokalaemia (flat T waves, u waves, ST segment depression) are indicators for giving intravenous potassium. A typical dose of 40 mmol/L potassium chloride in 1 L of 0.9% sodium chloride given at a rate of 10 mmol/hour is usually sufficient but local guidelines should be consulted. If potassium replacement is given it is mandatory to monitor the ECG throughout and for several hours after normokalaemia as there is a risk of rebound hyperkalaemia.
Key points
When assessing a myotonic disorder without obvious systemic involvement, do not forget myotonic dystrophy.
Symptoms of a muscle channelopathy are more than just weakness and myotonia: pain and fatigue can be prominent.
Andersen-Tawil syndrome requires cardiac monitoring regardless of symptoms.
Episodic movement disorders can be confused with a muscle channelopathy and vice versa.
Skeletal muscle channelopathies are treatable disorders.
Further reading
Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol 2015;5:761–90.
Jitpimolmard N, Matthews E, Fialho D. Treatment updates for neuromuscular channelopathies. Curr Treat Options Neurol 2020;22:34.
Matthews E, Balestrini S, Sisodiya SM, et al. Muscle and brain sodium channelopathies: genetic causes, clinical phenotypes, and management approaches. Lancet Child Adolesc Health 2020;4:536–47.
References
Footnotes
Twitter @EmmaM_Channels
Contributors All authors contributed to drafting and editing the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned: externally reviewed by Jon Walters, Swansea, UK.
Linked Articles
- Editors’ commentary
Other content recommended for you
- Genetic neurological channelopathies: molecular genetics and clinical phenotypes
- Diagnosis and new treatment in muscle channelopathies
- Muscle disease
- Neurological channelopathies
- Muscle diseases: mimics and chameleons
- Acute myotonic reaction during succinylcholine anaesthesia
- Ion channels and neurological disease: DNA based diagnosis is now possible, and ion channels may be important in common paroxysmal disorders
- Diverse phenotype of hypokalaemic periodic paralysis within a family
- 014 Prevalence study of skeletal muscle channelopathies in England
- Muscle weakness, palpitations and a small chin: the Andersen–Tawil syndrome