Article Text

Abstract
Corticobasal syndrome is a disorder of movement, cognition and behaviour with several possible underlying pathologies, including corticobasal degeneration. It presents insidiously and is slowly progressive. Clinicians should consider the diagnosis in people presenting with any combination of extrapyramidal features (with poor response to levodopa), apraxia or other parietal signs, aphasia and alien-limb phenomena. Neuroimaging showing asymmetrical perirolandic cortical changes supports the diagnosis, while advanced neuroimaging may give insight into the underlying pathology. Identifying corticobasal syndrome carries some management implications (especially if protein-based treatments arise in the future) and prognostic significance. Its treatment is largely symptomatic and is best undertaken within a multidisciplinary setting, including a neurologist, physiotherapist, occupational therapist, speech language therapist, psychiatrist and, ultimately, a palliative care clinician. Corticobasal syndrome can be a confusing entity for neurologists, not least because it has over time evolved from being considered predominantly as a movement disorder to a condition spanning a wide range of cognitive and motor manifestations. In this practical review, we attempt to disentangle this syndrome and provide clarity around diagnosis, its underlying pathological substrates, key clinical features and potential treatments.
- alien hand
- corticobasal degeneration
- apraxia
- aphasia
- myoclonus
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Case vignette 1 (with video)
A 68-year-old woman had a 2-year history of motor symptoms. Her first symptom had been her left hand not doing what it was told to do when drying the dishes. She also developed difficulties getting her words out. On examination, she had pseudobulbar speech and made dysphasic errors, and there was apraxia and hypometria of saccades, particularly leftward (video 1). She showed ideomotor apraxia and features of alien-limb syndrome in the left arm, and intermittent dystonic posturing of the left arm and leg but minimal limb rigidity. Her cognition was preserved.

Schematic of typical saccade abnormalities in CBS, compared with PD and PSP. In this schematic of eye movement recordings, patients were asked to make a leftward saccade of 20° towards a target as quickly as possible. Y axis is displacement amplitude, the X axis is time. Mild undershoot followed by a small secondary saccade is normal. Patients with PD commonly show mild hypometria (undershooting) requiring two or more corrective saccades to reach target. In CBS the degree of saccadic hypometria is often greater than in PD with the key feature being delayed launching of saccades (saccadic apraxia). In PSP the hallmark is early saccadic slowing (especially vertically) with considerable hypometria developing over time. CBS, corticobasal syndrome; PD, Parkinson’s disease; PSP, progressive supranuclear palsy. Figure by Bronstein & Anderson (2021), distributed at https://doi.org/10.6084/m9.figshare.14390951 under an open CC-BY 4.0 license.
We diagnosed corticobasal syndrome referred her to physiotherapy and occupational therapy. An MR scan of brain showed only mild involutional changes consistent with age but no perirolandic atrophy.
Her condition progressed over the next 4 years. She lost vertical eye movements and her alien limb became very pronounced. Her speech deteriorated to ‘yes’ and ‘no’, although she could still comprehend. She became more rigid with worsening dystonia particularly of neck extension, and her postural reflexes became impaired. We gave an unsuccessful trial of levodopa and sought speech and language involvement; botulinum injections into the neck extensors gave some benefit. She continued to deteriorate and died 6 years from symptom onset.
Case vignette 2
A 79-year-old woman reported decreased coordination, slowed movement and subtle right arm weakness that appeared to follow a fall. Over the next 9 months, her right arm became increasing difficult to use, causing difficulty with tasks such as doing up a bra and cutting food. Her walking felt more uncertain and she had one significant fall. On examination, there was marked right arm rigidity with a grasp reflex, significant bradykinesia and ideomotor apraxia. Eye movements were normal and there were no pyramidal nor cerebellar signs. Our impression was likely corticobasal syndrome. We arranged physiotherapy and occupational therapy, requested brain imaging and gave an empirical trial of levodopa.
Her right arm deficits progressed despite levodopa, which we later stopped. Her right arm became of little use to her, and she held it in a dystonic posture, without pain, though she still felt some agency over it. Her balance deteriorated further with frequent falls. She developed difficulty with speech, stumbling over longer words but her cognition remained unaffected. MR scan of brain showed left perirolandic atrophy consistent with corticobasal syndrome (figure 4). She remained at home with increasing support from physiotherapy and occupational therapy.
Historical context
In 1968, Rebeiz et al published three cases detailing the clinical and post mortem pathological findings of a ‘hitherto unrecognised disorder of the central nervous system’.1 All three had an asymmetric movement disorder characterised by slowed and awkward voluntary movements with additional involuntary movements. Pathological assessment identified frontoparietal atrophy driven by neuronal loss, gliosis and swelling of cell bodies, resulting in resistance to histological staining methods. While the cortex was primarily involved, the substantia nigra was abnormal in all three, and the dentatorubrothalamic system was abnormal in two. They coined the term ‘corticodentatonigral degeneration with neuronal achromasia’. Three decades later Gibb et al reported three further patients with similar clinical and histopathological findings. They adopted the shorter name ‘corticobasal degeneration’,2 and the next decade saw many further descriptions of this newly named disorder. The clinical phenotype expanded from primarily a movement disorder to include various cognitive and neurobehavioural deficits3–5 while the underlying pathology of clinically diagnosed cases also expanded to include Alzheimer’s disease, progressive supranuclear palsy (PSP), Pick’s disease and Creutzfeldt-Jakob disease.6–9 Thus, the etymology has slowly transitioned to ‘corticobasal syndrome’ as a clinical rather than a pathological diagnosis.10 Table 1 shows the current consensus diagnostic criteria for both the clinically defined corticobasal degeneration11 and the pathologically defined corticobasal syndrome.12 13 Figure 1 shows the common clinical phenotypes of corticobasal degeneration and the common pathologies underlying corticobasal syndrome.
Proposed criteria for corticobasal syndrome (the Cambridge criteria, modified Bak and Hodges)12

Unpicking corticobasal syndrome and corticobasal degeneration. From phenotype to underlying pathophysiology. This is a simplified view and includes only the common phenotypes of corticobasal degeneration and common pathological substrates underlying corticobasal syndrome. CBS, corticobasal syndrome; CBD, corticobasal disease; AD, Alzheimer’s disease; FTLD-TDP43, frontotemporal lobe degeneration TDP43; PSP, progressive supranuclear palsy; FTD Tau, frontotemporal dementia; FBSS, frontal behavioural-spatial syndrome; PPA, primary progressive aphasia.
Corticobasal degeneration
Corticobasal degeneration is a pathologically established four-repeat tauopathy.14 Its pathological features are cortical and striatal tau-positive neuronal and glial lesions of both white and grey matter, coupled with focal cortical and substantia nigra neuronal loss.14 Importantly, there is not a 1:1 mapping between corticobasal degeneration and corticobasal syndrome, and corticobasal degeneration pathology is associated with various clinical phenotypes (figure 1). There are four suggested broad clinical phenotypes:
Corticobasal syndrome.
Frontal behavioural-spatial syndrome.
Non-fluent/agrammatic variant of primary progressive aphasia.
PSP syndrome.11
Probable corticobasal degeneration criteria require an insidious onset and gradual progression for at least 1 year, age at onset >50 years, no similar family history or known tau mutations, and one of the clinical phenotypes outlined above. Features suggesting Parkinson’s disease (characteristic tremor, hallucinations, response to levodopa), or multiple system atrophy (prominent autonomic or cerebellar signs) are exclusions. However, the criteria still lack antemortem specificity to separate pathologically proven corticobasal degeneration from its mimics.15
Epidemiology of corticobasal syndrome
It is difficult to ascertain the true prevalence and incidence of corticobasal syndrome, given the varied use of the term and its interchangeability in early reviews with corticobasal degeneration. Estimates are therefore at best a guide and even then, remain crude. The estimated prevalence of corticobasal degeneration is 4.9–7.3 cases per 100 000 population.16 The annual incidence calculated from the prevalence and life expectancy would be between 0.5 and 1 per 100 000 per year, though this is higher than the rate observed in a population based study.17 The typical age of presentation is 50s–70s and average lifespan from diagnosis to death is 7 years. There does not appear to be any sex bias.18
Genetics of corticobasal syndrome
A single pathogenic mutation is unlikely to contribute greatly to the pathogenesis of corticobasal syndrome. However, familial clustering can occur with up to 31% have a family history of parkinsonism or dementia19 The most common monogenic mutations associated with familial corticobasal syndrome are in microtubule-associated protein tau (MAPT) resulting in frontotemporal lobar degeneration (FTLD)-tau pathology strongly resembling corticobasal degeneration,20 although genome-wide association studies have identified other single nucleotide polymorphisms.21 More recently corticobasal syndrome has been associated with FTLD with ubiquitin-immunoreactive inclusions (FTLD)22 or TAR DNA-binding protein 43 (TDP-43) leading to frontal temporal lobe degeneration (FTLD-TDP)23 both of which are most often caused by progranulin mutations24 but not always.25 Pathogenic GGGCC expansion with mutations in C9orf72 (chromosome 9 open reading frame 72) and mutations in LRRK2 (previously limited to Parkinson’s disease)26 are also associated with corticobasal syndrome.27 Outside of familial monogenic mutations, a case–control study suggests single-nucleotide polymorphisms in the H1 haplotype of the MAPT gene may predispose to sporadic corticobasal syndrome.28
Clinical signs and symptoms in corticobasal syndrome
Corticobasal syndrome has an insidious onset and is slowly progressive.12 29 Patients with dramatic presentations and/or rapidly progressive disease courses should be considered mimics (see below).
Motor features
Extrapyramidal motor features are common with no dramatic or sustained response to levodopa therapy.12 29 Rigidity is the most frequent extrapyramidal motor sign, present in 73%–100% of cases, mostly presenting as an asymmetric akinetic–rigid syndrome.13 29 30 Dystonia is much less common than rigidity and tends to affect a single limb, often the upper and usually early in the disease course.12 13 Other extrapyramidal features such as bradykinesia and postural instability may also occur.12 A tremor can develop but is an action or postural jerky movement that subsides with rest, and is quite unlike a resting Parkinson’s disease tremor.12 13 29 It can overlap with another common motor feature—myoclonus—which occurs in roughly 40% of cases.12 13 29 Electrophysiology studies suggest the myoclonus is cortical or subcortical in origin.31–33
Alien limb syndrome
The alien limb syndrome comprises involuntary limb movements combined with an altered sense of limb belonging or ownership. It usually involves the hand but may uncommonly occur only in the leg, or both arm and leg, and rarely is bilateral.34 35 A detailed account of the underlying neurobiological processes causing alien limb is beyond this review but proposed mechanisms have been suggested.36 The alien limb is easily confused with other neurological signs (table 2). There are three recognised variants: frontal, callosal (together termed ‘anterior’) and posterior (figure 2).
Alien limb differential diagnosis

Classification algorithm of the alien limb syndrome. Modified from Hassan and Josephs. 72 CBS, corticobasal syndrome; CJD, Creutzfeldt-Jakob disease.
The posterior variant is the most often encountered type in corticobasal syndrome and usually affects the non-dominant upper limb, with lesions involving the non-dominant parietal lobe.35 It is characterised by a sense that the affected limb does not belong to the person. There are usually other parietal cortical deficits including sensory hemineglect, and astereognosis. The typical motor features are not as intrusive as in the frontal variant but may take the form of levitation and other non-purposeful actions, abnormal posturing and ataxia.
Corticobasal syndrome is easily the most common cause of an alien limb (two-thirds of cases).35 By the same token the alien limb syndrome develops in about a half of people with corticobasal syndrome.35 37 While the asymmetry of corticobasal syndrome involves the left and right hemispheres equally, alien limb in this condition usually develops in the non-dominant limb, for unclear reasons.36 In patients presenting with alien limb, the timing of onset during the disease may help to suggest the cause; for example, it can be the presenting symptom of Creutzfeldt-Jakob disease but occurs a median of 1 year after disease onset in corticobasal syndrome.35 The associated neurology can also help in the differential diagnosis. Thus, mirror movements develop in 40% of corticobasal syndrome patients with the alien limb but are uncommon in other causes, while intermanual conflict is very uncommon in corticobasal syndrome.36 Myoclonus is usual in patients with Creutzfeldt-Jakob disease but common in corticobasal syndrome, and uncommon in other causes of alien limb.35
There are no proven treatments for alien limb syndrome and management approaches are based on anecdotal experience and the type of alien limb. The frontal variant may respond to sensory tricks (eg, wearing a glove), distracting tasks (eg, holding a ball in the hand), verbal cues that enhance voluntary action and cognitive–behavioural therapy for anxiety reduction. For the posterior variant (common in corticobasal syndrome), treatments used have included clonazepam, botulinum toxin injections into the most active proximal muscles, visualisation strategies (eg, putting the affected hand into a mirror box) and spatial recognition tasks, but these approaches are not always well tolerated or maintained and there is scant information on their long-term benefits.38
Apraxia
Limb apraxia is among the most commonly identified signs that suggests cortical dysfunction in the corticobasal syndrome, occurring in 70%–80%.39–41 Apraxia is defined as a disorder of higher level motor control, manifesting as impaired skilled and learnt motor acts, despite intact primary sensory and motor pathways39 Apraxia generally affects both sides of the body. Because corticobasal syndrome is usually asymmetrical, finding apraxia on the less affected side (as is common) adds weight to the conclusion that abnormality of movements are not simply due to extrapyramidal features such as rigidity and bradykinesia.40
When screening patients for the presence of apraxia, it can help to test different types of complex movements—thought to correspond to different underlying neurobiological processes that can be disrupted by brain pathology.39 These include:
Performing a gesture, miming the use of tools and copying meaningless gestures. Deficits in these movements, usually referred to as ideomotor apraxia, are common in corticobasal syndrome and can be readily assessed in the clinic.
Performing complex, multistep tasks. Deficits in these processes are often referred to as conceptual, or ideational apraxias, capturing the idea that it is loss of knowledge about objects and their associated actions that underlies the patient’s difficulties. This is harder to screen for in a routine clinic appointment but may be inferred from the history, or from a formal occupational therapy assessment. Ideational apraxia can be extremely disabling for a person’s day-to-day functioning.
Performing repetitive distal limb movements such as tapping the thumb with each finger in turn. Deficits such as clumsy or inaccurate movements, are referred to as limb-kinetic apraxia—a somewhat controversial classification that can be difficult to distinguish from the effects of weakness or bradykinesia.
Other sorts of higher order cortical dysfunction are often termed apraxias—for example, gait apraxia, constructional apraxia, dressing apraxia, orobuccal apraxia and apraxia of speech (to name a few). When present, these point towards cortical dysfunction signs that can provide evidence for the presence of a corticobasal syndrome.
Apraxia of saccades and other eye movements in corticobasal syndrome
The traditional oculomotor hallmark of clinically diagnosed corticobasal syndrome is saccade apraxia,42 43 which manifests clinically as difficulty and delay in initiating saccades towards a target, usually with the use of an assisting simultaneous or preceding head movement, and in the laboratory as a substantial increase in saccade latency.44 45 Typically, the saccadic apraxia is greatest towards the side with the greatest limb apraxia.42 43 In contrast to PSP, saccade velocities in patients with corticobasal syndrome are normal46 47 (figure 3). Smooth pursuit can also be moderately impaired but not as severely as in patients with PSP. The neuropathological substrate of saccadic apraxia in corticobasal syndrome awaits further clarification but it remains a distinctly useful clinical diagnostic feature.
Aphasia
The typical language disturbance in corticobasal syndrome is non-fluent variant primary progressive aphasia, with slowed, effortful and/or groping (apraxia of speech) speech and grammatical errors being common.48 However, patients can also develop a logopenic aphasia, characterised by prominent difficulty in word retrieval and sentence repetition.48 The latter is commonly associated with underlying Alzheimer’s disease pathology, while non-fluent variant primary progressive aphasia, including apraxia of speech, may suggest tau pathology. Therefore, aside from providing evidence of ‘cortical’ involvement, the pattern of aphasia may help to identify the pathology underlying corticobasal syndrome, but further research is required.
Neuropsychiatric aspects of corticobasal syndrome
Corticobasal syndrome has a range of neuropsychiatric comorbidities. However, the lack of large scale studies means that while we commonly see features such as depression, apathy, anxiety and agitation (among others) in the clinic, we do not have accurate estimates of their prevalence at different stages of corticobasal syndrome—or know whether these features associate with particular underlying pathologies.49 A study of 15 patients with what we would now refer to as corticobasal syndrome found particularly high rates of depression and apathy and also an absence of hallucinations.50 This latter point suggests that the presence of visual hallucinations in a patient with parkinsonism and cognitive deficits should raise concerns they may in fact have an alpha-synucleinopathy such as dementia with Lewy bodies.
We advocate for screening all patients presenting with corticobasal syndrome for neuropsychiatric symptoms, particularly as these features have a major impact on quality of life for patients and their families. Screening can be performed formally (eg, using the neuropsychiatric inventory,51 or by questioning both the patient and an informant for presence of mood disturbance (dysphoria, anhedonia, anxiety), behavioural change (apathy, obsessive or compulsive behaviours, agitation, irritability, impulsivity, loss of empathy) and psychotic features (hallucinations, delusions). Education of caregivers about the possibility of emergence of these complications can be helpful.
Neuroimaging in corticobasal syndrome
Brain imaging has three roles in the assessment of patients with corticobasal syndrome—ruling out mimics/structural causes (see below), providing support for the clinical diagnosis of a corticobasal syndrome, and providing clues to the underlying pathology.33
Imaging correlates
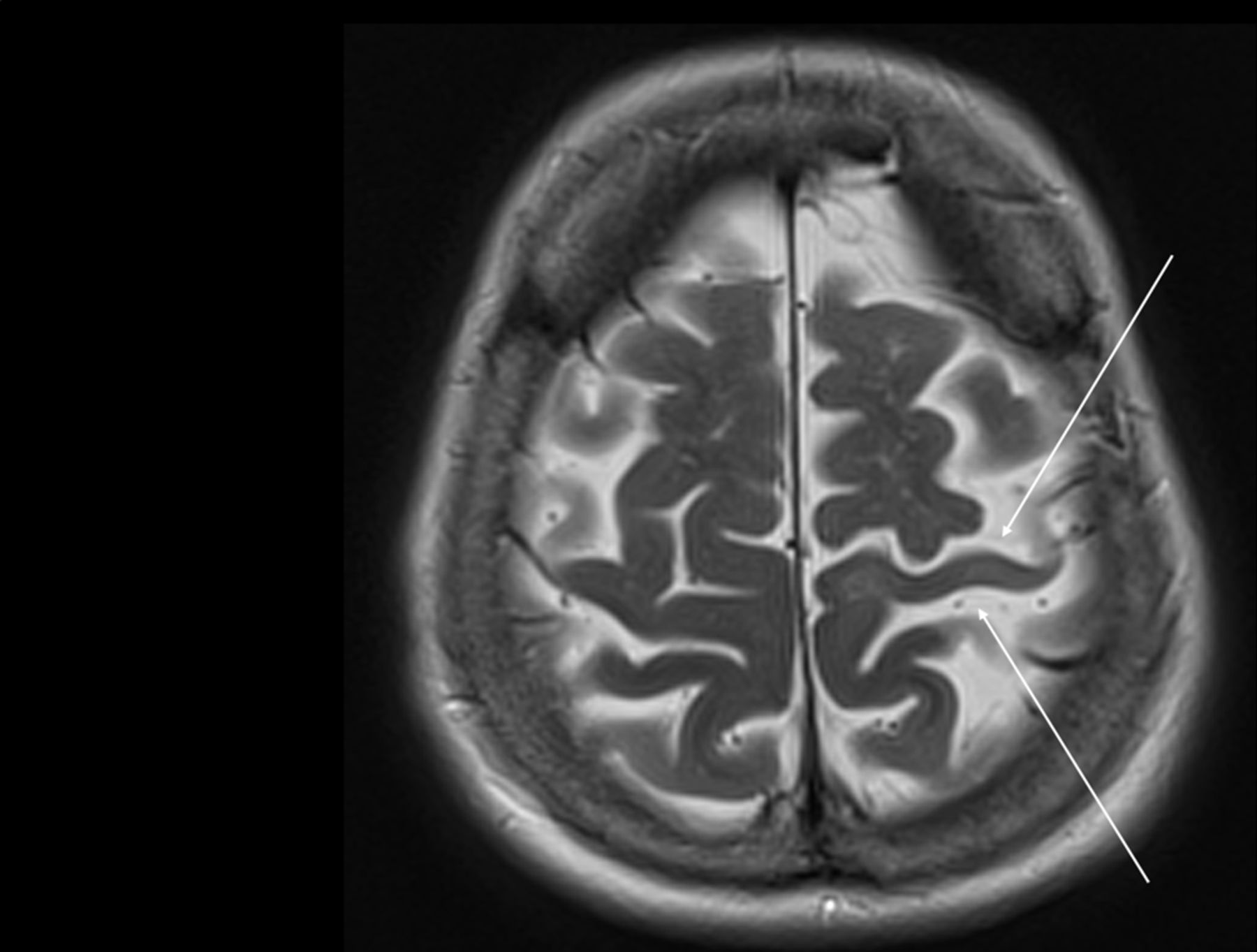
Corticobasal syndrome is associated with asymmetrical cortical changes in markers of neuronal loss or dysfunction (grey matter atrophy, hypometabolism or hypoperfusion). This particularly affects frontal-parietal regions encompassing premotor, motor and sensory association cortex, and typically develop contralateral to the more affected side of the body33 52 53 (figure 4). Notably, such ‘perirolandic’ patterns of change do not appear specific to any underlying pathology but instead associate directly with the clinical features of corticobasal syndrome, consistent with the importance of these regions for processing higher order sensory information and translating this into motor actions. Therefore, finding asymmetrical perirolandic atrophy or hypometabolism on clinical imaging supports a clinical diagnosis of corticobasal syndrome, though its absence does not exclude it. This can be particularly helpful early in the disease course, when the differential may include Parkinson’s disease or other parkinsonian syndromes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Example of an MR scan of a brain in a patient with corticobasal syndrome A. Small arrows show moderate focal asymmetric left perirolandic atrophy on T2-weighted imaging.
Striatal dopamine transporter (DAT) density can be imaged and measured using single-photon emission CT or positron-emission tomography (PET). Most, but not all, patients with corticobasal syndrome have a positive DAT scan.54 One follow-up study suggests that in time all such patients will have a positive result.55 For the clinician, the DAT scan’s limitation is that it does not differentiate corticobasal syndrome from other parkinsonian disorders.
Imaging to predict the underlying pathology
There has been a recent emphasis on developing measures to identify reliably the underlying pathology in corticobasal syndrome.53 Such measures will be increasingly relevant as protein-specific treatments hopefully emerge over the coming years.33 Ultimately their utility will depend on their ability to distinguish between pathologies at individual rather than at group level.
These strategies can be split into techniques that identify the presence of abnormal protein (such as imaging to detect increased concentrations of brain amyloid protein), and techniques that identify patterns—either in neuronal loss/metabolism or brain connectivity—closely associated with the underlying pathology. The use of amyloid PET imaging to identify corticobasal syndrome caused by Alzheimer’s pathology is the clearest example of the former approach. In turn, researchers are now examining clinical and standard imaging correlates of amyloid positive and negative groups to further refine understanding of how corticobasal syndrome may differ between pathologies.56 Given that there is often an associated underlying tauopathy, emerging tau-based PET techniques—which are still troubled by some technical issues such as off-target binding—are also generating strong interest for their potential to identify underlying corticobasal degeneration or progressive supranuclear pathology.33 57
Finally, the distribution of neuronal loss or brain hypometabolism in patients with corticobasal syndrome predicts the underlying pathology, at least at a group level. In particular, corticobasal syndrome caused by Alzheimer’s pathology often has a posterior pattern of hypometabolism, while corticobasal degeneration may show more subcortical hypometabolism, and PSP pathology shows more frontal hypometabolism.52 58 More complicated techniques assessing brain structural (white matter) or functional connectivity are also showing promise for distinguishing between pathologies but are not yet clinically useful.59
In summary, a corticobasal syndrome diagnosis can be supported—but not refuted—by imaging features, while emerging techniques may direct the neurologist to the underlying pathological cause of a patient’s syndrome—information that over time will have practical relevance.
Treatment of corticobasal syndrome
There are currently no proven treatments for corticobasal syndrome. Recent advances in the treatment of tauopathies with immunotherapies and gene expression show promise,60 61 but for the moment we emphasise the importance of making a diagnosis that can explain a puzzling array of problems for a patient and their family. It provides a valid explanation for their symptoms and allows a reframing of priorities from obtaining a diagnosis to coping with the problem. Ideally, treatment should be provided within a multidisciplinary setting with expertise provided by a neurologist, physiotherapist, occupational therapist, speech language therapist, psychiatrist and, ultimately, palliative care services.
Although parkinsonism in corticobasal syndrome does not generally respond well to levodopa, most patients will try it as part of their initial assessments (often when the diagnosis is less clear), and it is reasonable to push the dose up towards at least 1000 mg/day before classifying a patient as a ‘non-responder’.62 In our experience, other dopaminergic therapies (dopamine agonists, monoamine oxidase inhibitors) also have very limited efficacy in treating motor symptoms of corticobasal syndrome, but a dopamine agonist may be worth considering in those with prominent apathy. Options for treating troublesome myoclonus include levetiracetam and clonazepam. Dystonia can be functionally disabling and at times painful. Anticholinergics, benzodiazepines and amantadine provide modest help at best, and adverse effects—especially cognitive impairment, hallucinations and confusion—often outweigh any benefit, particularly in older patients, while amantadine can also cause insomnia and leg oedema.63 Botulinum toxin injections can help, depending on the dystonic pattern. Particularly when treating upper limb dystonia, the disabling effects of symptoms must be weighed against the potential limb weakness resulting from injections—but as with other interventions a pragmatic trial is certainly reasonable.37 Physiotherapy input is also important in optimising mobility following botulinum toxin injections.
Specific options to consider for the alien limb syndrome are summarised above. Management to mitigate the effects of apraxia is best coordinated by an occupational therapist with knowledge of the condition. Speech therapists can teach patients techniques to overcome some of their language deficits and it is worth seeking their input when speech difficulties are a prominent feature—they can also provide patients with practical advice if swallowing difficulties develop. We often refer patients for physiotherapy aimed at strength and balance training as well as gait assessment—the addition of gait aids can allow some people to maintain relative physical independence. Although there are no proven treatments for cognitive deficits such as memory and attentional impairment in corticobasal syndrome, many clinicians consider trialling cholinesterase inhibitors if there is a strong suggestion from the history (memory impairment), examination (predominate cortical signs) and cognitive assessment (visuospatial or memory deficits) to suggest an underlying Alzheimer’s disease pathology.
There are several available pharmacological options for neuropsychiatric manifestations.62 Many mild behavioural issues may be better managed non-pharmacologically (caregiver education, environment changes, etc) but undoubtedly medications can help with more severe disruptions. Seeking psychiatric guidance is useful and building a strong relationship with an interested psychiatrist can help patient management and improve job satisfaction. Selective serotonin reuptake inhibitors are useful for treating common problems such as anxiety, depression and obsessive-compulsive disorder. Apathy, or reduced motivated behaviour, is common, debilitating and difficult to treat. Informing caregivers that it is part of the disease process can help. Agents targeting dopaminergic, cholinergic and serotonergic neuromodulatory networks may help apathy in other degenerative disorders, but there is no good evidence to guide use of these treatments in corticobasal syndrome. Finally, some patients will develop marked behavioural disturbances, including irritability/aggression and psychosis. Management can be difficult but can include atypical antipsychotics, for example, quetiapine or clozapine with appropriate blood count monitoring.
Other practical issues to address include driving safety and checking whether driving licensing agencies need to be informed, the importance of updating a person’s will, and establishing an enduring power of attorney early in the disease course, as these can be problematic later if significant cognitive impairment develops. Lastly, putting patients in touch with local charities can greatly help patients and families. If specific charities are not available (eg, the PSP association in the UK and curePSP in the USA) exploring Parkinson’s and dementia charities is a reasonable first step.
Corticobasal syndrome mimics
Conditions that may initially be diagnosed as corticobasal syndrome but turn out to be something else tend to be those with subacute or chronic onset, and those that have some, but not all, symptoms and signs resembling true neurodegenerative corticobasal syndrome. Most of these mimics feature alien limb syndrome with or without myoclonus, and/or one of the rapidly progressive dementias, often with aphasia. Thus, many of the non-neurodegenerative causes of the alien limb syndrome may mimic corticobasal syndrome, including stiff-person syndrome,64 Hashimoto’s encephalitis,65 66 thalamic cavernoma,67 as well as Creutzfeldt-Jakob disease68 and other rapidly progressive dementias.69 Mimics can usually be distinguished from true corticobasal syndrome by the careful neurological examination to identify peripheral (such as areflexia, proprioceptive loss) or central (eg, pyramidal) nervous system signs, combined with appropriate investigations such as MR scan of brain, electroencephalogram, serum antineuronal and other autoantibody assays, which together may indicate an alternative diagnosis. It is critical to identify these mimics as early as possible as many are treatable or have a better prognosis than true corticobasal syndrome. A careful family history is also important, and clinicians should have a low threshold for genetic testing especially in younger patients with atypical features.
Prognosis
The prognosis for a patient diagnosed with corticobasal syndrome depends mainly on the underlying neuropathology (i.e. cause), the difficulty being that that cause is not easily determined during life. Consequently, there is little available information to assist counselling of the patient and family. In a study of 10 Japanese patients with corticobasal syndrome coming to post mortem (three each with corticobasal degeneration, PSP and Alzheimer’s disease pathology, and one with atypical tauopathy) median survival was 7 years with a range of 4–15 years. Survival was similar across pathologies.70 An earlier study of 14 patients with pathologically confirmed corticobasal degeneration reported a median survival time after onset of symptoms of 7.9 years with a considerable range of 2.5–12.5 years. Survival was shorter in those with early and widespread parkinsonism or frontal lobe syndrome.71 In summary, on present knowledge, average survival in corticobasal syndrome is 7–8 years but with a considerable range of some 3–15 years.
Conclusion
Corticobasal syndrome is a disorder of movement, cognition and behaviour, caused by several underlying pathologies including corticobasal degeneration. Clinicians should consider the diagnosis in patients presenting with any combination of extrapyramidal features, apraxia or other parietal signs, aphasia and alien limb phenomena. Neuroimaging showing asymmetrical perirolandic cortical changes supports the diagnosis and advanced neuroimaging may give insight into the underlying pathology. We suggest neuropsychological screening in all patients presenting with corticobasal syndrome. Identifying corticobasal syndrome carries some prognostic significance, management implications and in the future if protein-based treatments arise—may direct further investigations as to underlying pathology.
Key points
Corticobasal syndrome is a clinical entity with many different underlying pathologies, including corticobasal degeneration.
Corticobasal degeneration is a pathological diagnosis associated with several clinical syndromes, one of which is corticobasal syndrome.
Corticobasal syndrome has a varied presentation: distinguishing clinical features include asymmetric parkinsonism, myoclonus, alien limb, cortical sensory loss, eye and limb apraxia, and imaging may show asymmetric perirolandic atrophy.
Corticobasal syndrome has several important mimics (eg, Creutzfeldt-Jakob disease, Hashimoto’s encephalitis), some of which are treatable.
Further reading
Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80(5):496–503. doi: 10.1212/WNL.0b013e31827f0fd1
Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psych 2012;83(4):405–10. doi: 10.1136/jnnp-2011-300875
Pardini M, Huey ED, Spina S, et al. FDG-PET patterns associated with underlying pathology in corticobasal syndrome. Neurology 2019;92(10):e1121–35. doi: 10.1212/WNL.0000000000007038
Ethics statements
References
Footnotes
Contributors DW, CLH and TA all contributed equally to the manuscript. All were involved in manuscript conception, drafting and critical revisions.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned. Externally peer reviewed by Negin Holland, Cambridge, UK and Oliver Bandmann, Sheffield, UK.
Linked Articles
- Editors’ commentary
Other content recommended for you
- Neuropathology and emerging biomarkers in corticobasal syndrome
- Predictors of survival in frontotemporal lobar degeneration syndromes
- Pathology of neurodegenerative disease for the general neurologist
- Clinicoradiological and neuropathological evaluation of primary progressive aphasia
- Predicting loss of independence and mortality in frontotemporal lobar degeneration syndromes
- Therapeutic trial design for frontotemporal dementia and related disorders
- Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review
- Validation of the new consensus criteria for the diagnosis of corticobasal degeneration
- Diagnostic criteria for corticobasal syndrome: a comparative study
- Globular glial tauopathy type II