Article Text

Abstract
The early and accurate diagnosis of dementia is more important than ever before but remains challenging. Dementia is increasingly the business of neurologists and, with ageing populations worldwide, will become even more so in future. Here we outline a practical, symptom-led, bedside approach to suspecting dementia and its likely diagnosis, inspired by clinical experience and based on recognition of characteristic syndromic patterns. We show how clinical intuition reflects underlying signature profiles of brain involvement by the diseases that cause dementia and suggest next steps that can be taken to define the diagnosis. We propose ‘canaries’ that provide an early warning signal of emerging dementia and highlight the ‘chameleons’ that disguise or mimic this, as well as the ‘zebras’ that herald a rare (and sometimes curable) diagnostic opportunity.
- dementia
- alzheimer disease
- aphasia
- cognition
- lewy body
Statistics from Altmetric.com
Introduction
As the number of people worldwide with dementia approaches 50 million, the need for early and accurate diagnosis is more urgent than ever.1 Timely diagnosis avoids the limbo of diagnostic uncertainty and futile cycles of investigation, equips patients and families to engage appropriate support and to plan for the future, and directs rational and appropriate management.2 It will also be essential for the effective deployment of disease-modifying therapies that are on the horizon. However, the early diagnosis of dementia is challenging and remains peculiarly reliant on clinical judgement (box 1); the target diseases are complex and affect aspects of higher brain function that are generally not assessed in routine neurological practice. Treatises on dementia conventionally list the clinical features of particular diseases—in the trenches, however, the biggest challenge is often suspecting dementia in the first place and deciding why this is not ‘just’ Alzheimer’s disease.
Some principles of bedside cognitive assessment
History taking
History is the most important aspect of successful dementia diagnosis.
Obtaining a history from reliable informants who know the patient well is integral and interviewing them separately may encourage sharing of sensitive or embarrassing clues to the diagnosis.
A minute or two spent putting the patient and family at ease is well invested.
How organised and detailed patients seem when describing their symptoms is informative, particularly if at odds with performance on formal cognitive tests.
Interpretation of cognitive or behavioural changes depends on an appreciation of the patient’s sociocultural background, education, occupation, premorbid language skills, any pre-existing specific developmental or other deficits, and medical and psychiatric history (including medications).
Cognitive concerns will most frequently be framed as a non-specific ‘memory’ problem: this is the most ubiquitous of several potential ‘pitfall’ symptoms that must be deconstructed (see table 2).
Domains of cognitive function and behaviour that may not be volunteered should also be explored (as these help define the cognitive profile), framing these as questions about functioning in daily life.
Particularly in younger people, a detailed family history is essential (including parents’ diagnoses and age at death if relevant, and the ages of any siblings).
Examination
It is first essential to establish that the patient is alert and cooperative and that their peripheral vision and hearing are adequate (or corrected as appropriate).
Observing the patient’s conduct and interaction with the examiner and others is often telling (it may point to frontal lobe dysfunction more clearly than any test; see table 2).
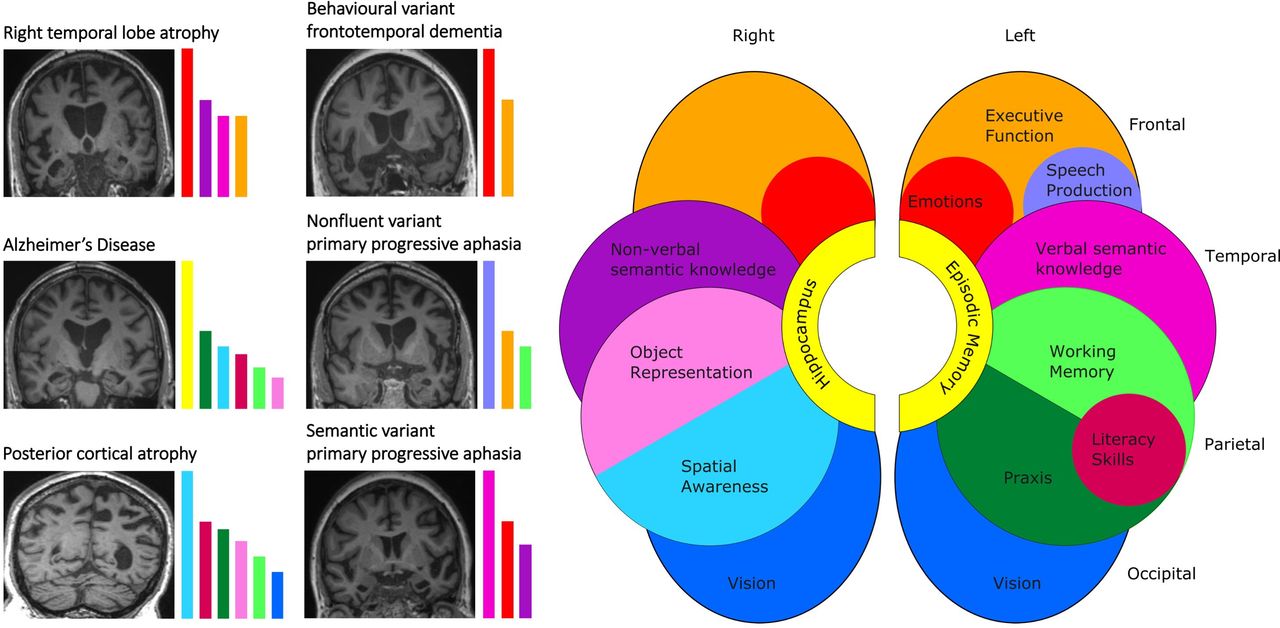
To corroborate the history and to build a diagnostic profile of cognitive deficits, it is helpful to have a scheme for testing cognition (a ‘walk around the brain’, table 1), armed with some tools to elicit cognitive deficits (figure 1): the cognitive profile in turn predicts the underlying pattern of brain involvement (figure 2).
Quantitative cognitive assessments such as the Mini-Mental State Examination, the Montreal Cognitive Assessment and Addenbrooke’s Cognitive Examination are widely available; however, each has its limitations1 33 and none in itself should be used to diagnose or exclude dementia.
The general neurological and systemic examinations are essential, particularly for substantiating diagnoses other than Alzheimer’s disease (see tables 3 and 4).
Some noteworthy potential ‘pitfall’ symptoms and signs requiring further clarification or interpretation in suspected dementia
Dementia is a syndrome that can be defined very generally as a progressive decline in cognitive function and/or behaviour that impacts daily life functioning. As such, it has a multiplicity of causes. Most of these are neurodegenerative pathologies that are not presently reversible; however, the rare exceptions are not to be missed. A key theme in dementia (especially in neurodegenerative disease) is that the causative pathologies initially target certain brain functions relatively selectively, due to a predilection of pathogenic proteins to involve particular brain networks.3 Over time, these signature patterns become obscured as the spread of pathological change leads to convergent, widespread damage and impairment. The window of greatest opportunity for accurate diagnosis (and anticipated interventions) is therefore early-stage disease. Appreciating how profiles of brain damage relate to cognitive deficits is key to deciding which diseases are likely in patients presenting with suspected dementia.
Here we outline a symptom-led, bedside approach to suspecting dementia that we have found useful in busy neurological clinics. First, we consider clues that help one decide whether or not cognitive decline is present and, if so, the likely cause. We show how these clues predictably reflect underlying signature patterns of brain involvement by causative pathologies and suggest next steps that can be taken to define the diagnosis. As with many other disorders, the neurologist’s essential task is to identify ‘canaries’ that provide an early warning signal of emerging disease, avoid being misled by ‘chameleons’ that disguise or mimic this and remain alert to the occasional ‘zebra’ that heralds a rare (and sometimes curable) diagnosis.
We suggest some general principles and tools for cognitive assessment in box 1 and figure 1 and outline a bedside framework for cognitive history taking and examination in table 1. Diagnostic canaries based on characteristic patterns of cerebral involvement are listed in table 1; potential pitfalls are listed in table 2, chameleons in table 3 and zebras in table 4.

Examples of aids for testing cognition at the bedside. These are some examples of materials that we use to sample a range of cognitive domains (see also table 1 and figure 2) when examining a patient with suspected dementia (shown with permission of Professor E K Warrington). (A) A pictorial scene for description, to assess connected language (fluency, word finding and expressive grammar) and ability to parse a complex environment (visuospatial processing). (B) An array of pictures of familiar items for assessing word retrieval (naming), single-word comprehension (pointing to an item named by the examiner) and subsequent item recall (anterograde episodic memory). (C) A passage for reading aloud to elicit difficulties sounding non-words (personal names, eg, ‘Scorba’; phonological dyslexia) or irregular words (eg, ‘yacht’, regularised as ‘yached’; surface dyslexia) or with tracking lines of text (visuospatial processing). (D) Fragmented letters to assess visual apperceptive function. (E) A Stroop task, requiring the patient to state the (conflicting) colour of the ink in which each colour name is printed (in fluent readers of English, a test of response inhibition—one aspect of executive function).
Bedside assessment of the patient with suspected dementia: a ‘walk around the cognitive brain’
Some important ‘chameleons’ of dementia diagnosis
Some important ‘zebras’ in dementia diagnosis
Does this patient have dementia?
Distinguishing early dementia from the ‘worried well’ or a ‘functional’ cognitive disorder is an increasingly frequent challenge faced by neurologists as public awareness and anxiety about dementia continue to increase.4 A functional cognitive disorder should be considered if there are positive features of internal inconsistency, that is, ability to perform a task well at certain times, but with significant difficulty when it becomes the focus of attention. The person who gives a detailed (or even overinclusive) account of their memory lapses, attributes their difficulties eloquently to specific past events and who is substantially more concerned about their cognitive function than their partner, children or colleagues—often attending clinic unaccompanied—is more likely to be anxious or to have a functional cognitive disorder than dementia. People with obsessional personalities are more prone to overinterpret the imperfections of normal memory. There is often a flavour of wavering concentration, such as being unable to remember why one has entered a room, misplacing household items in odd locations (eg, keys in the fridge) or ‘going blank’ during a conversation only to have the required information re-emerge soon afterward. Cognitive testing frequently generates considerable anxiety, inducing ‘thought-blocking’ and performance may vary widely between assessments, often leading to marked inconsistencies (disastrous test scores despite evident competence in daily life). This contrasts with the ‘face-saving’, humour and minimisation often seen in Alzheimer’s disease, or indifference in diseases where insight is impaired. However, particularly in older patients, functional cognitive impairment may signal an emerging neurodegenerative process which declares itself subsequently.
Depression or other primary psychiatric diagnoses must not be overlooked—these are potentially treatable and undetected carry significant risk of harm. There is often a history of previous psychiatric episodes, though this may not be volunteered. Core depressive symptoms are low mood or anhedonia, variably accompanied by fatigue, psychomotor retardation, impaired concentration, a sense of personal worthlessness, significant change in appetite or recurrent morbid thoughts.5 Depressed patients are often downcast and disengaged, giving frequent ‘don’t know’ responses. Active psychosis also leads to poor engagement, and there may be evidence of delusional thinking or verbal hallucinations. It is important to keep in mind that anxiety, mood changes and psychosis occur not uncommonly in ‘organic’ dementias and may be early features6 7; moreover, organic deficits may be elaborated by patients with abnormal illness behaviour, and certain syndromes (such as parietal presentations of Alzheimer’s disease and behavioural variant frontotemporal dementia) are notoriously prone to psychiatric mislabelling even by experienced clinicians.
Some important canaries
Alzheimer’s disease
Older patients with episodic and topographical memory impairment that declines over time will most commonly have emerging Alzheimer’s disease. Details of important events and conversations are not retained, questions become repetitive and there is often a loss of facility with route-finding and a history of becoming lost. There may be a signal ‘catastrophic’ episode (often, disorientation in unfamiliar surroundings, e.g. while on holiday) preceding more pervasive deterioration. Difficulty following conversations in background noise and dislike of noisy environments (due to impaired auditory scene processing) tend to develop early.8 Loss of pleasure in reading (probably multifactorial in nature) is also frequently reported. Retained (partial) awareness of limitations as well as endogenous emotional changes wrought by the disease often lead to loss of confidence or initiative, embarrassment, anxiety and withdrawal from social activities, manifesting in the clinic as a tendency to ‘trail’ the accompanying person into the room and to turn to them (‘head-turning sign’) when asked a question.6 9 On bedside testing, the extent to which cognitive impairment has been masked by a well-preserved social façade may be surprising: knowledge of current affairs tends to be vague, recall of previously presented items does not benefit from cueing and topographical material (such as reconstructing the journey to the hospital) may be notably affected.
Beyond memory, parietal cortical functions including word retrieval, praxis, calculation and visuospatial function are affected relatively early in Alzheimer’s disease. There may be difficulties with word finding, handling household appliances, managing money or visuospatially demanding activities such as driving or do-it-yourself. Coming to grips with new technology taxes learning, executive and parietal functions and is often particularly challenging.
Dementia with Lewy bodies
Dementia commonly develops in Parkinson’s disease (particularly in older patients) and is a core feature of Lewy body pathology.10 Clues include an early predisposition to severe, prolonged delirium (for example, attending a minor infection or surgical procedure) and sometimes acting out of dream content (REM sleep behaviour disorder, due to loss of normal skeletal muscle atonia). Later, misperceptions and hallucinations (usually predominantly visual) develop, though these may not be volunteered. Initially, they may be brief transients glimpsed in the periphery but evolve into vivid, animate entities (commonly faces, people or animals) that emerge out of background features (such as foliage or a pile of clothes) in stereotyped fashion, particularly under low-light conditions. In contrast to psychotic or dopamine-driven hallucinations, these are typically non-threatening though insight into their nature may be impaired. An ‘extracampine’ sense of a presence beyond the field of vision is common. Marked fluctuations in alertness, attention and cognitive competence even within the course of a day (particularly deterioration later in the day) are also characteristic.11 Complaints of ‘double vision’ and problems reading (without identifiable ocular pathology) are common, and difficulty using gadgets such as a smartphone exposes executive and parietal dysfunction. Features of associated Parkinsonism (such as hypomimia or gait changes) may be subtle initially but it is worth asking about autonomic symptoms (particularly urinary urgency, nocturia or unexplained collapses).12 13
Vascular cognitive impairment
The most common cognitive syndrome of cerebrovascular disease is not the stepwise decline in function of classical teaching, but an insidious deterioration characterised by disorganisation, loss of verve and initiative, irritability, mental rigidity, emotional lability and other mood changes, and sometimes inappropriate or disinhibited social behaviour. Vascular risk factors are common, but their absence does not exclude the diagnosis. Examination typically reveals cognitive and affective blunting, executive dysfunction, impaired attention and recall (which in contrast to Alzheimer’s disease, does generally benefit from cueing) with variable additional, more focal deficits;14 15 however these may be over-estimated due to often significant slowing of mentation. Brisk tendon and pout reflexes (despite flexor plantar responses) where present are characteristic and in more advanced cases gait may be wide-based and shuffling. Dysphagia may develop later in the disease. Though this is one of the few diagnoses in neurology where a compatible scan is sine qua non, over-diagnosis of incidental vascular changes is a significant pitfall, compounded by the frequent coexistence of cerebrovascular with primary neurodegenerative pathologies.
Some chameleons and zebras
Alzheimer’s disease variants
Atypical presentations of Alzheimer’s disease dominated by non-amnestic deficits are not uncommon, particularly in younger people16 17; conversely, Alzheimer’s disease is simulated by a variety of other disease processes.18 Table 3 summarises some of these chameleons; it is particularly important to consider potentially reversible mimics, such as transient epileptic amnesia or obstructive sleep apnoea.18–24 There are three major Alzheimer variant syndromes, likely reflecting differential involvement of the same core temporo–parieto–frontal brain network targeted by Alzheimer pathology (see tables 1 and 2, and figure 2).16–18 These variants lie on a clinical continuum and overlap is frequent.
{kind=link}
{kind=link}
Cognitive and neuroanatomical profiles of some major dementia syndromes. The cartoon brain (right) shows cognitive functions predominantly mediated by different cerebral regions in each cerebral hemisphere. The T1-weighted coronal MRI sections (left) show characteristic patterns of regional brain atrophy in representative neurodegenerative disorders (typical Alzheimer’s disease—disproportionate, bilateral hippocampal atrophy; behavioural variant frontotemporal dementia—predominant bilateral but asymmetric, frontal and anterior temporal lobe atrophy; non-fluent/agrammatic variant primary progressive aphasia—predominantly left-sided, anterior peri-Sylvian cortical atrophy; posterior cortical atrophy—predominant biparietal atrophy; semantic variant primary progressive aphasia—selective, predominantly left-sided anteromesial and inferior temporal lobe atrophy; right temporal lobe atrophy—selective, predominantly right-sided anterior temporal lobe atrophy). In each case, the dementia syndrome reflects a profile of brain network breakdown (see also table 1); this correspondence is indicated by the coloured histograms alongside each scan, which code the cognitive domains pre-eminently affected in each syndrome. The histogram colours follow the same convention as the brain cartoon; the heights of the histogram bars are arbitrary but indicate the relative clinical prominence of deficits across cognitive domains that are typically seen in a particular syndrome. The left hemisphere is projected on the right (following standard clinical convention) throughout.
Posterior cortical atrophy, the ‘visual variant’ of Alzheimer’s disease, usually presents with impairments of visuospatial awareness, reading and praxis.17 Beware the patient who has made numerous futile visits to the optician or who describes more difficulty reading pixelated signs than newsprint (signifying visual apperceptive agnosia): this is the cardinal degenerative disorder of the visual brain, disrupting the interpretation of visual scenes despite normal sensory acuity. There is usually a full hand of accompanying parietal lobe deficits, but episodic memory early on is often well preserved.
Logopenic aphasia is the language-led variant of Alzheimer’s disease within the primary progressive aphasia spectrum.25 It is characterised by prominent word-finding difficulty, conversational pauses (sentences tend to trail off) with speech sound (phonological) errors and, on examination, impaired repetition of phrases despite intact repetition of single words (due to reduced verbal working memory).26
The ‘frontal’ (behavioural/dysexecutive) variant of Alzheimer’s disease remains the least well defined.27 Clinically, it can closely resemble the behavioural variant of frontotemporal dementia, but prominent accompanying memory impairment and confabulation may be bedside clues to the diagnosis.
Frontotemporal dementia syndromes
Among the ‘zebras’ of dementia diagnosis, the frontotemporal dementias are particularly important because they are collectively a major cause of dementia in middle life and wreak havoc on social and occupational functioning. This is a diverse group of diseases with complex neurobiology28; however, three major clinical presentations are recognised.
The behavioural variant of frontotemporal dementia presents with abnormalities of social and emotional awareness and reactivity.28 29 The patient generally lacks insight, but the family complains bitterly that they have ‘changed’, typically with loss of warmth and social skills (there may have been embarrassing faux pas), and frequently prominent apathy, rituals and/or impulsivity that may have resulted in loss of a job or ill-advised decisions. Gluttony and development of a pathological sweet tooth are characteristic, exemplifying a much broader repertoire of odd, inflexible and maladaptive behaviours with valuation of abstract or impersonal interests over other people. These features may be particularly striking in patients with selective right temporal lobe atrophy,30 who also frequently exhibit prosopagnosia. Behavioural variant frontotemporal dementia is challenging to diagnose, particularly early on, as there are few reliable biomarkers. Patients may do well on formal cognitive (including executive) tests. There are several highly pertinent clinical issues surrounding the diagnosis: it is genetically mediated in up to perhaps a third of cases (genetic testing for the three major causative, autosomal dominant mutations should be considered in all younger patients) and vigilant neurological follow-up is indicated, both to detect the emergence of major associations (atypical parkinsonism or motor neurone disease) and to identify patients who fail to manifest abnormalities on brain MRI or metabolic (fluorodeoxyglucose - positron emission tomography (FDG-PET) or single-photon emission computed tomography (SPECT)) imaging. The nosological status of these latter ‘phenocopy’ cases is still unclear.
Among language-led dementia syndromes (the primary progressive aphasias), the non-fluent/agrammatic variant is the most immediately clinically striking. These patients characteristically have effortful, unmelodious, misarticulated ‘apraxic’ speech and their utterances may be terse and agrammatic (‘telegraphic’). Early on, there may be particular difficulty with public speaking, reversing of ‘yes’ and ‘no’ or re-emergence of a childhood stutter. Initially, naming and comprehension are largely intact and written expression is usually more fluent than speech.25 26 As the syndrome evolves, impairments of orofacial praxis (affecting volitional movements such as whistling) and dysphagia often supervene, frequently with emergence of an extrapyramidal syndrome in the corticobasal—progressive supranuclear palsy spectrum.
In contrast, the semantic variant of primary progressive aphasia presents with increasingly circumlocutory and vacant speech that is well constructed and fluent (even garrulous). These patients characteristically have asked family members the meanings of words (‘What’s a tornado?’) and often compile personal ‘dictionaries’. They have early, profound anomia, underpinned by impaired single-word comprehension and vocabulary loss affecting all language channels, often extending to a tendency to sound irregular words (such as ‘sew’) as they are printed (‘surface dyslexia’25 26). The true nature of this syndrome is captured in its older designation, ‘semantic dementia’: this is the paradigmatic disorder of the semantic memory system that mediates knowledge about words, objects and concepts. As it evolves, non-verbal semantic knowledge about visual and other sensory objects and about the emotional and social signals of other people also disintegrates. Patients generally develop a behavioural syndrome similar to behavioural variant frontotemporal dementia. In our experience, there is invariably focal, asymmetric, usually predominantly left-sided anterior temporal lobe atrophy on brain MRI at presentation (figure 2); if this is absent, we hesitate to invoke the diagnosis.
Other ‘zebras’
These are many and diverse (table 4): clues to their presence include younger age of onset, a family history of younger onset dementia (often labelled as Alzheimer’s disease or psychiatric illness), prominent extracognitive neurological or systemic features or a rapid course. The last group includes catastrophic illnesses such as prion disease but also several reversible processes that demand careful exclusion (table 4). Diagnosis of the many rare diseases that cause dementia in younger adults due to metabolic, inflammatory, storage and other inherited disorders generally depends on clinical features or markers beyond cognition31 32: these disorders tend to produce a fairly nondescript ‘frontosubcortical’ cognitive syndrome led by executive and neuropsychiatric dysfunction.
Next steps after suspecting dementia
If available, more detailed cognitive testing with a neuropsychologist is often a valuable extension to the bedside assessment to: quantify suspected deficits in relation to age-appropriate norms and premorbid attainment; detect deficits in domains (such as executive function) that are challenging to assess at the bedside; and compare performance over serial assessments, which may be diagnostic in cases of clinical doubt.
Any patient with suspected dementia should have brain imaging (ideally MRI)—occasionally this will show a surgically remediable process but more generally it defines the profile of atrophy in neurodegenerative diseases (figure 2) and detects signal alterations such as those associated with cerebrovascular disease, leukodystrophies and prion disease. Serial imaging of change over a year or more can be informative if the first scan is normal. Conversely, frontal or parietal ‘atrophy’ is quite commonly overinterpreted on MRI. Brain FDG-PET or SPECT is sometimes useful to demonstrate regional cerebral hypometabolism where Alzheimer’s disease or a frontotemporal dementia is suspected but MRI is inconclusive.
Although basic haematological and metabolic screens are worthwhile to detect potentially reversible factors that may contribute to cognitive decline, these are rarely the primary culprit. Diagnostic markers of dementia are currently largely derived from CSF analysis, which should be considered in anyone with younger onset dementia (arbitrarily, before the age of 65 years) or rapid evolution, when it is likely to have the most useful predictive value (see tables 3 and 4)—relevant CSF constituents include cells and oligoclonal bands (pointers to brain inflammation), neurofilament light chain (a non-specific indicator of the presence and severity of neuronal damage) and more specific protein profiles of Alzheimer pathology (raised total and phospho-tau, elevated tau:beta-amyloid42 or beta-amyloid40:amyloid42 ratio).
Diagnostic testing for causative genetic mutations should be considered in younger patients where there is a compatible phenotype and in particular a suggestive (autosomal dominant) family history, but only after appropriately informed counselling in the clinic, particularly with respect to the implications for other family members. Other more specialised investigations may be appropriate in certain clinical contexts (see table 4).
Local services should be engaged for support early and people with atypical forms of Alzheimer's disease or non-Alzheimer dementias can be directed to Rare Dementia Support (https://www.raredementiasupport.org/).
Conclusions
Early and accurate diagnosis of dementia is desirable and achievable, but it must first be suspected. As always in neurology, pattern recognition is key. The first challenge is to determine whether dementia is likely and then, based on a functionally oriented history and systematic examination, to determine the profile of cerebral involvement and thus the candidate underlying pathology. Despite a growing array of ancillary tools, clinical judgement is likely to remain essential and indeed, to assume even greater importance as effective treatments become available.
Key points
Dementia is a syndrome of progressive decline in cognitive function and/or behaviour that impacts upon daily life functioning and has multiple potential causes.
Timely diagnosis is desirable and achievable with a systematic approach to cognitive assessment.
The major dementias target particular brain networks and accordingly have distinct phenotypes.
The diagnosis rests primarily on clinical assessment, with neuropsychological, neuroimaging and biomarker support where appropriate.
Further reading
McWhirter L, Ritchie C, Stone J, et al. Functional cognitive disorders: a systematic review. Lancet Psychiatry 2020;7:191–207.Oxford Textbook of Cognitive Neurology and Dementia. Husain M, Schott JM, editors. Oxford University Press; 2016.
Crutch SJ, Lehmann M, Schott JM, et al. Posterior cortical atrophy. Lancet Neurol 2012;11:170–8.
Sivasathiaseelan H, Marshall CR, Agustus JL, et al. Frontotemporal dementia: a clinical review. Semin Neurol 2019;39:251–63.
Ethics statements
Acknowledgments
The Dementia Research Centre is supported by Alzheimer's Research UK, Brain Research UK, and the Wolfson Foundation. This work was supported by the Alzheimer’s Society, Alzheimer’s Research UK and the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
References
Footnotes
Twitter @cjdhardy, @charl_marshall, @ftdtalk, @jmschott, @rimonaweil
Contributors JCSJ and JDW conceived of and drafted the main manuscript. All authors contributed significantly to the content of the manuscript and reviewed the drafts and provided feedback.
Funding JCSJ is supported by an Association of British Neurologists Clinical Research Training Fellowship, funded by Guarantors of Brain. CJDH is supported by a RNID-Dunhill Medical Trust Pauline Ashley Fellowship (PA23). SJC receives support from the ESRC/NIHR Dementia Research Initiative (ES/S010467/1). CRM is supported by a grant from Bart’s Charity. RSW is supported by a Wellcome Clinical Research Career Development Fellowship (201567/Z/16/Z). LMcW receives philanthropic funding from Baillie Gifford. JDW receives grant support from Alzheimer’s Research UK, the Alzheimer’s Society and the National Brain Appeal (Frontotemporal Dementia Research Studentship in Memory of David Blechner).
Competing interests None declared.
Provenance and peer review Commissioned. Externally peer reviewed by Anthony Bayer, Cardiff, UK.
Linked Articles
- Editors’ commentary
- Editors’ commentary
Other content recommended for you
- Cerebrospinal fluid biomarkers in the differential diagnosis of Alzheimer's disease from other cortical dementias
- Lifting the veil: how to use clinical neuropsychology to assess dementia
- Alzheimer's disease: mimics and chameleons
- Utility of testing for apraxia and associated features in dementia
- Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review
- Neuroimaging in dementia: a practical guide
- Apraxia screening predicts Alzheimer pathology in frontotemporal dementia
- Theory of mind in behavioural-variant frontotemporal dementia and Alzheimer's disease: a meta-analysis
- Biomarkers in dementia: clinical utility and new directions
- Which neuropsychiatric and behavioural features distinguish frontal and temporal variants of frontotemporal dementia from Alzheimer's disease?