Article Text

Abstract
Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) typically presents in middle life with a combination of neuropathy, ataxia and vestibular disease, with patients reporting progressive imbalance, oscillopsia, sensory disturbance and a dry cough. Examination identifies a sensory neuropathy or neuronopathy and bilaterally impaired vestibulo-ocular reflex. The underlying genetic basis is of biallelic AAGGG expansions in the second intron of replication factor complex subunit 1 (RFC1). The frequency and phenotype spectrum of RFC1 disease is expanding, ranging from typical CANVAS to site-restricted variants affecting the sensory nerves, cerebellum and/or the vestibular system. Given the wide phenotype spectrum of RFC1, the differential diagnosis is broad. RFC1 disease due to biallelic AAGGG expansions is probably the most common cause of recessive ataxia. The key to suspecting the disease (and prompt genetic testing) is a thorough clinical examination assessing the three affected systems and noting the presence of chronic cough.
- cerebellar ataxia
- neuropathy
- neurogenetics
Statistics from Altmetric.com
First clinical descriptions of the cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS)
The first description of the clinical syndrome of combined vestibular, neuropathic and cerebellar dysfunction with absent vestibular function and reduced smooth-pursuit optokinetic eye movements was in 1991.1 In 2004, Migliaccio et al reported four patients with the syndrome of cerebellar ataxia with bilateral vestibulopathy.2 The term CANVAS was coined in 2011 when the same group reported 23 additional patients and showed that a length-dependent sensory deficit is an integral feature of this syndrome.3 Later, some patients with this condition were identified as having autonomic dysfunction and a dry spasmodic cough.4 5
Pathological studies in typical CANVAS cases have found loss of Purkinje cells, predominantly in the vermis of the cerebellum, and a marked loss of sensory neurones in the dorsal root and V, VII and VIII cranial nerve ganglia.6 7 Although there were few postmortem studies, they helpfully localised the sensory impairment of CANVAS to the spinal and cranial sensory ganglia, and the vestibular impairment to the vestibular ganglia.
Identification replication factor complex subunit 1 (RFC1) expansion causing CANVAS, late-onset ataxia and sensory neuro(no)pathy
Using a combined recessive linkage and whole-genome sequencing approach, we identified biallelic AAGGG expansions in the second intron of RFC1 in familial and sporadic CANVAS and late-onset ataxia cases.8
This novel repeat expansion associated with CANVAS has since been confirmed by multiple groups and in patients of differing ethnicities.8–15
More recently, novel pathogenic (AAAGG)10–25(AAGGG)exp(AAAGG)4–6 and (ACAGG)exp configurations were identified in people from Oceania and East Asia.16–18 Despite the clinical similarities between CANVAS and multiple system atrophy, RFC1 expansions were either absent or very rare in people with multiple system atrophy of Caucasian or Chinese Han origin; this further supports its specificity to CANVAS and its spectrum of disease.19–22
The mechanism underlying neurodegeneration in RFC1 CANVAS is still unknown. The RFC1 gene is implicated in DNA replication and repair, but preliminary studies have not shown either reduced expression or overt loss of function of RFC1 protein.8 11
Clinical features of RFC1 disease spectrum
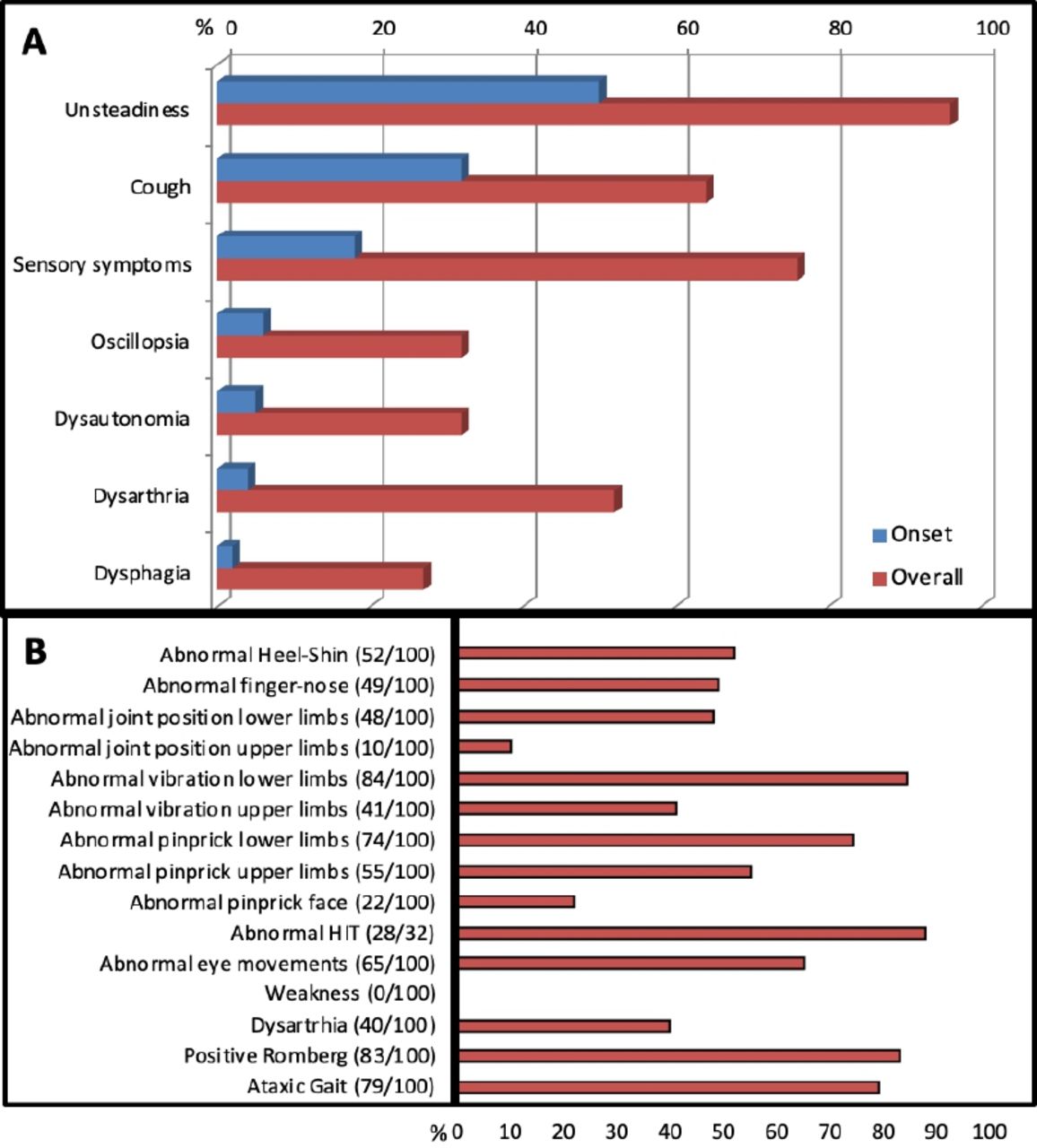
We recently described the clinical features in 100 genetically confirmed cases with biallelic RFC1 expansions.23 Cases were enrolled from multiple centres from the UK, Europe, South America, Australia and New Zealand. Figure 1 details their history and examination findings. The mean age of onset of neurological manifestations was 52 (range 19–76) years, and the mean age at the time of study was 72 years.
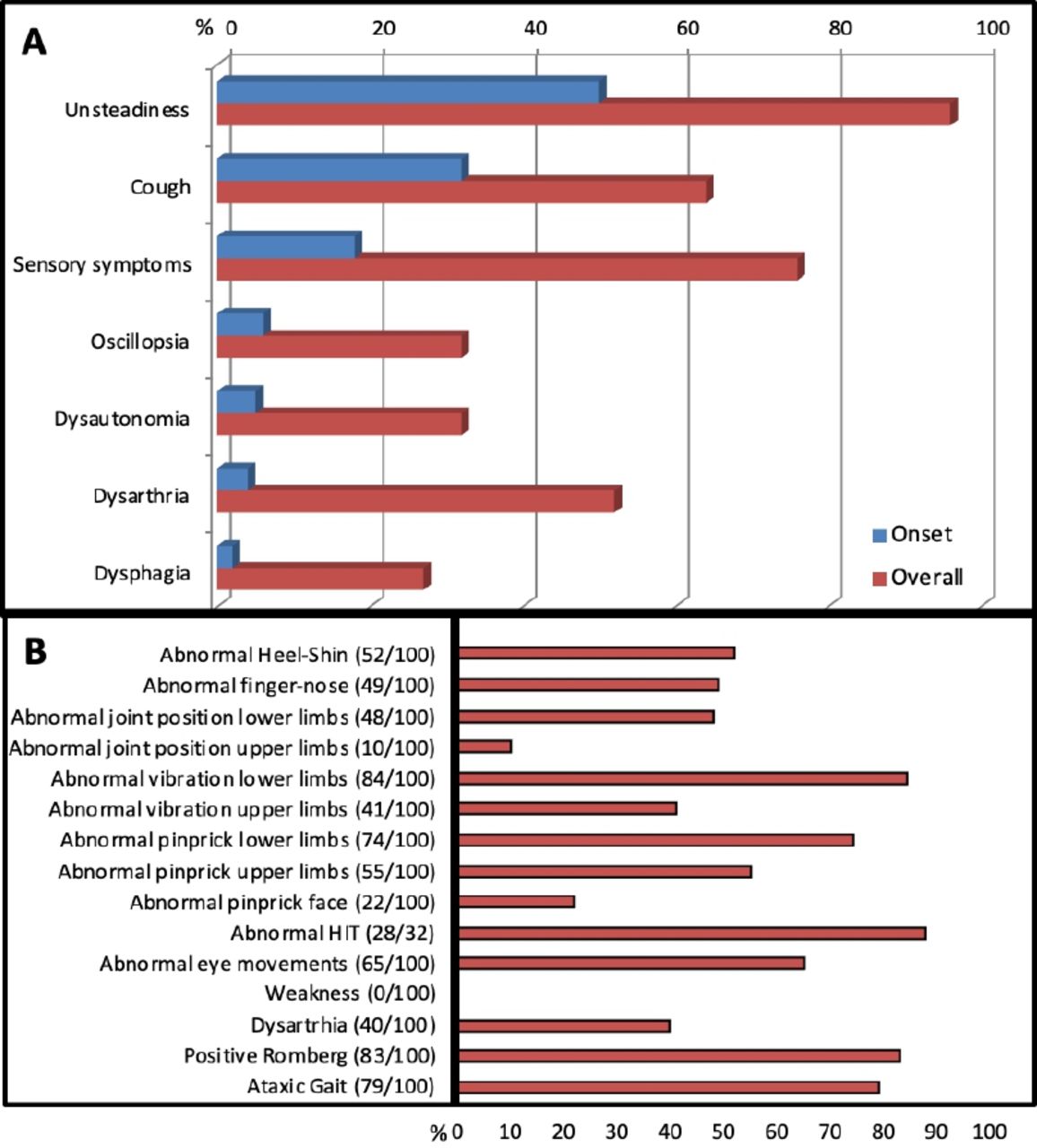
Clinical features of genetically confirmed replication factor complex subunit 1 cerebellar ataxia, neuropathy and vestibular areflexia syndrome and its disease spectrum. (A) Symptoms at onset and during disease progression. (B) Detailed neurological examination. HIT, head-impulse test.
Progressive imbalance was the most common complaint and the first symptom in half of cases. The imbalance was often worse in the dark, suggesting a prominent peripheral large-fibre sensory component.
Over two-thirds had sensory symptoms, including loss of feeling, pins and needles and neuropathic pain. Neurological examination identified impaired sensation to pinprick, vibration and joint position, usually in a length-dependent distribution. Reflexes were reduced/absent, retained or even brisk, but muscle strength was always normal.
One-third of cases reported oscillopsia, defined as a visual disturbance where objects appear to oscillate during head movements; this is a common result of a bilaterally impaired vestibulo-ocular reflex. Conversely, vertigo, defined as an abnormal sensation of motion in which the individual or their surroundings seem to whirl dizzily, and usually stemming from an acute/subacute and generally unilateral impairment of the vestibular system, was rare in RFC1 CANVAS and, when reported, was probably unrelated. Up to 90% of cases showed evidence of bilateral vestibular hypofunction on bedside head-impulse testing. Previous studies have also shown that impaired visually enhanced vestibulo-ocular reflexes are common and indicate the coexistence of vestibular and cerebellar pathology.2
Dysarthria and dysphagia, probably attributable to cerebellar dysfunction, was a frequent complication in later stages. However abnormal eye movements of putative cerebellar origin were present earlier in the disease course, commonly including gaze-evoked, downbeat and horizontal nystagmus, broken pursuits and dysmetric saccades.
Two-thirds of cases reported a chronic spasmodic dry cough; this appeared as early as in the second decade of life, and up to three decades before any neurological symptom develops. About half of cases reported autonomic dysfunction, including postural hypotension, erectile dysfunction, chronic constipation, urinary dysfunction and altered sweating, but this was rarely disabling.
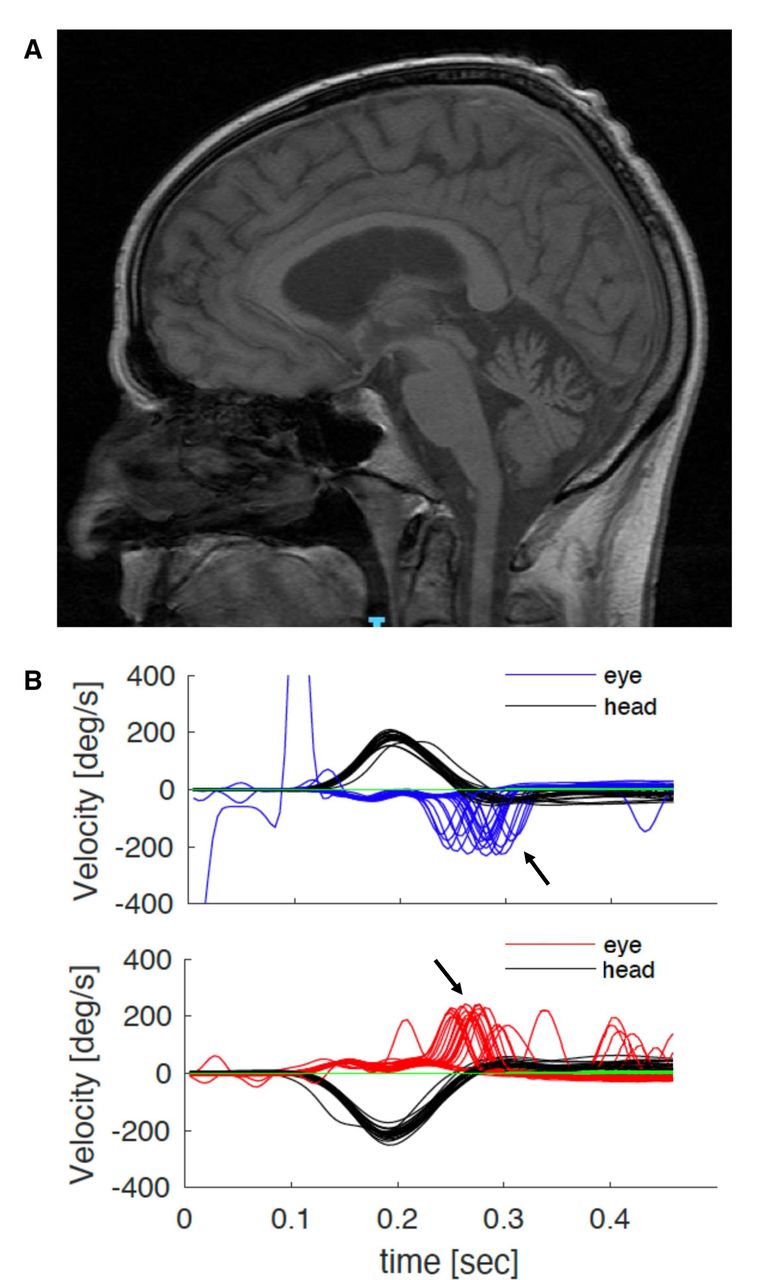
Magnetic resonance (MR) scan of the brain showed cerebellar atrophy, particularly of the vermis, in ~60%; vestibular testing was abnormal in over 90% of cases tested (figure 2), while nerve conduction studies universally showed a sensory neuropathy. The sensory action potentials were reduced or absent; motor studies were normal in 90%, and 10% showed minor reductions in motor action potentials and/or distal denervation. Blink and masseter reflexes can become impaired as a result of the widespread sensory neuronopathy, which also involves the trigeminal nerves.5 The H reflex, where integrity implies a normal function of both peripheral motor fibres and muscle spindle afferent fibres, was normal when assessed by a different group in a small series of patients with CANVAS; this explains the frequent occurrence of normoreflexia or hyper-reflexia.5 Autonomic testing identified parasympathetic and/or sympathetic dysfunction in half of our series of genetically confirmed cases. Finally, peripheral nerve ultrasound scanning showed a reduced cross-sectional area of upper and lower limb nerves, suggesting a role for this test in clinically defined CANVAS.24 25
{kind=link}
{kind=link}
Investigations in typical RFC1 CANVAS. (A) MR scan of the brain of a patient with genetically confirmed RFC1 CANVAS showing parenchymal volume loss affecting the cerebellum, both within the cerebellar hemispheres and the cerebellar vermis. (B) Video of the head-impulse test. Head velocity (black lines) and eye velocity traces for head impulses rightward (top panel) and leftward (bottom panel). The eye traces show the compensatory saccades (catch-up saccades, arrows) at the end of the head movement. CANVAS, cerebellar ataxia, neuropathy and vestibular areflexia syndrome; RFC1, replication factor complex subunit 1.
The disease shows a slowly progressive course. Half of the patients needed a stick when walking after 10 years and a quarter were wheelchair-dependent 5 years later. However, life expectancy was not affected.
Two-thirds of genetically confirmed cases had full CANVAS after clinical examination and investigations. None had isolated cerebellar syndrome or bilateral vestibular failure, but in 15 cases, the only clinically detectable manifestation of the disease was a sensory neuropathy.
Although the natural history of CANVAS awaits fuller investigation, our retrospective data suggest a pattern of spatial progression from the early involvement of sensory neurones to the vestibular system and later the cerebellum.
Box 1 illustrates a typical case of RFC1 CANVAS. The routine evaluation of any patient with ataxia and/or sensory neuro(no)pathy should include enquiry about unexplained cough and a thorough clinical examination, including bedside head-impulse testing looking for bilateral vestibular areflexia.
Illustrative case of RFC1 CANVAS
Case study: a man with progressive imbalance and distal pins and needles
A man in his early 50s had a 3-year history of dizziness and imbalance, particularly when walking on uneven surfaces and in the dark, along with loss of feeling and pins and needles in his feet. He had no weakness, muscle cramps or symptoms of dysautonomia. His symptoms had slowly progressed and had worsened in the last year. He had a history of a dry chronic spasmodic cough since his 20s.
There was no family history of neurological disease.
He had a slightly broad-based ataxic gait. He could not walk heel to toe. Romberg’s test was positive. There was gaze-evoked horizontal nystagmus and broken slow-pursuit eye movements. His head-impulse test identified a bilaterally impaired vestibular ocular reflex indicating a bilateral vestibular areflexia, with an abnormal, visually enhanced vestibulo-ocular reflex, pointing to combined vestibular and cerebellar impairment. Muscle tone, bulk and strength were normal. Reflexes were all present and plantars were downgoing. Sensory examination showed reduced pinprick to the knees and the wrists, reduced vibration to the costal margin but normal position sense.
Nerve conduction studies found normal motor studies with absent sensory action potentials in the upper and lower limbs. MR scan of the brain showed mild cerebellar vermis atrophy (figure 2A). MR scan of the spine showed atrophy and T2 hyperintensity in the posterior columns. Video head-impulse test confirmed bilateral vestibular areflexia (figure 2B). Relevant blood tests including serum B12, folate, glycated haemoglobin, immunofixation and paraneoplastic autoantibodies were negative. RFC1 testing confirmed a biallelic AAGGG expansion.
Signs suggesting RFC1 CANVAS
Progressive sensory ataxic neuropathy.*
Chronic cough.
Altered vestibular ocular reflex and visually enhanced vestibulo-ocular reflex.
Gaze-evoked nystagmus, broken pursuits.
Dysarthria and dysphagia (more advanced stages of the disease).
Atypical signs, making RFC1 CANVAS less likely
Absence of sensory neuropathy.
Presence of motor involvement.
Neurological onset in the first decade of life.
Rapid progression.
Prominent dysautonomia.
*Consider RFC1 testing in anyone with unexplained sensory neuropathy after negative laboratory screening for acquired causes.44
CANVAS, cerebellar ataxia, neuropathy and vestibular areflexia syndrome; MR, magnetic resonance; RFC1, replication factor complex subunit 1.
Differential diagnosis
Given the complex phenotype of RFC1 CANVAS, the differential diagnosis includes genetic and acquired causes of neuropathy, ataxia and vestibular disease. Table 1 summarises the key differential diagnoses.26–42
Differential diagnoses of replication factor complex subunit 1 cerebellar ataxia, neuropathy and vestibular areflexia syndrome and its disease spectrum
The main differential diagnosis in patients with late-onset cerebellar ataxia is multiple system atrophy, a rapidly progressive neurodegenerative disease. The features suggesting multiple system atrophy rather than RFC1 disease include rapid progression,28 early and severe autonomic involvement, absence of sensory neuropathy and vestibular dysfunction, the association with rapid eye movement sleep behaviour disorder, parkinsonism, and the typical MR brain scan pattern with putaminal, pontine, and middle cerebellar peduncle atrophy and 'hot cross-bun' signs cruciform T2 hyperintensity in the pons.21 29
Friedreich’s ataxia shows remarkable similarities to CANVAS. Both are caused by recessive repeat expansions and lead to progressive degeneration of sensory neurones, accompanied by cerebellar dysfunction. Vestibular areflexia can also develop in Friedreich’s ataxia. However, unlike CANVAS, Friedreich’s ataxia typically begins before the age of 25 years. Also, muscle atrophy, spasticity and skeletal deformities (pes cavus, scoliosis) are frequent in Friedreich’s ataxia but absent in CANVAS. Cardiomyopathy, diabetes mellitus, vision and hearing involvement also often complicate the disease course of Friedreich’s ataxia.30–32 Among other differential genetic causes, we recently showed that the phenotype spectrum of the dominant R199C mutation in RNF170, formerly identified in Eastern Canadian patients with posterior column ataxia,26 encompasses autosomal dominant sensory ganglionopathy and vestibular areflexia and should be considered in RFC1 negative CANVAS cases, particularly with dominant family history and normal cerebellar function.27
Several mitochondrial diseases due to nuclear or mitochondrial genetic defects can also manifest with ataxia, neuropathy and, more occasionally with bilateral vestibular areflexia. In particular, a biallelic mutation in polymerase gamma associated with sensory ataxic neuropathy, dysarthria and ophthalmoplegia should be considered in the diagnostic work-up of someone with progressive sensory ataxic neuropathy. Clues to suspect a mitochondrial disease are an earlier age of onset, the frequent association of chronic progressive external ophthalmoplegia and the multisystem involvement.42
Genetic testing for RFC1 expansions
The molecular diagnosis of RFC1 CANVAS relies on a bespoke testing entailing flanking and repeat-primed PCR. Given the large size of the AAGGG expansions, ranging from several hundreds to several thousand repeated units, the only way to confirm the presence of biallelic repeat expansions and their size is by laborious and time-consuming Southern blotting.
Note that the currently available next-generation sequencing gene panels do not detect RFC1 expansions. Other techniques, including long-read sequencing,43 may have the potential to assess the presence, size and sequence of repeat expansions in RFC1 reliably and may be a valuable alternative to the current multistep algorithm.
Due to its genetic complexities, RFC1 CANVAS genetic testing is available in the UK only as a research test, although work is in progress to introduce it into routine diagnostic testing.
Conclusions
Since its identification, the frequency and phenotype spectrum of RFC1 disease have expanded, ranging from typical CANVAS, where it explains over 90% of cases, to site-restricted variants affecting prominently or exclusively the sensory nerves, the cerebellum or the vestibular system. Given that up to 8% of healthy chromosomes carry the AAGGG expansion across different ethnicities—which is higher than FXN GAA expansion associated with Friedreich’s ataxia (allele frequency ~1%)—RFC1 disease due to biallelic AAGGG expansions is probably the most common cause of recessive ataxia. The key to diagnosis is a thorough clinical examination assessing the three affected systems, as well as identifying the presence of chronic cough, and prompt genetic testing.
Key points
Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) typically presents in middle life with progressive imbalance, oscillopsia, sensory disturbance and a dry cough; examination typically identifies a sensory neuropathy or neuronopathy and bilateral impairment of the vestibulo-ocular reflex.
The underlying genetic basis is of biallelic AAGGG expansions in the second intron of replication factor complex subunit 1 (RFC1).
Enquiring about the presence of cough and routine assessment of eye movements (nystagmus, broken pursuits) and bedside head-impulse test in all cases of sensory neuropathy are key to suspecting RFC1 disease and prompt genetic testing.
Further reading
Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 2019;51:649–58.
Cortese A, Tozza S, Yau WY, Rossi S, Beecroft SJ, Jaunmuktane Z, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 2020;143:480–90.
Currò R, Salvalaggio A, Tozza S, et al. RFC1 expansions are a common cause of idiopathic sensory neuropathy. Brain 2021;144:1542–50.
Ethics statements
Patient consent for publication
Acknowledgments
We thank Dr Silvia Colnaghi for kindly providing the video head-impulse test traces showed in figure 2B.
RC is supported by the European Academy of Neurology (EAN) Research Fellowship 2021.
References
Footnotes
Contributors AC, MMR and HH designed the study. AC, RC, EV and WYY drafted the manuscript. All authors revised and approved the manuscript.
Funding AC thanks the Medical Research Council (MRC) (MR/T001712/1), the Fondazione CARIPLO (2019–1836), the Italian Ministry of Health Ricerca Corrente 2018–2019 and 2020, the Inherited Neuropathy Consortium and the Fondazione Regionale per la Ricerca Biomedica for grant support. HH and MMR thank the MRC, the Wellcome Trust, the MDA, MD UK, Ataxia UK, The MSA Trust, the Rosetrees Trust and the NIHR UCLH BRC for grant support.
Competing interests None declared.
Provenance and peer review Provenance and peer review. Commissioned. Externally peer reviewed by Simon Hammans, Southampton, UK.
Other content recommended for you
- Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS): diagnostic contribution of vestibular function tests
- Diagnostic yield of testing for RFC1 repeat expansions in patients with unexplained adult-onset cerebellar ataxia
- Sensory neuronopathies: new genes, new antibodies and new concepts
- Can CANVAS due to RFC1 biallelic expansions present with pure ataxia?
- RFC1-related ataxia is a mimic of early multiple system atrophy
- Intronic FGF14 GAA repeat expansions are a common cause of ataxia syndromes with neuropathy and bilateral vestibulopathy
- Diagnosis and management of progressive ataxia in adults
- RFC1 CANVAS: the expanding phenotype
- Bilateral vestibulopathy presaging clinically probable multisystem atrophy
- 15.45 Recessive pentanucleotide repeat expansion in RFC1 causes CANVAS and late-onset sensory ataxia