Article Text

Abstract
Autoinflammatory syndromes result from a defective innate immune system. They are characterised by unexplained fever and systemic inflammation involving the skin, muscle, joints, serosa and eyes, along with elevated acute phase reactants. Autoinflammatory syndromes are increasingly recognised as a cause of neurological disease with a diverse range of manifestations. Corticosteroids, colchicine and targeted therapies are effective if started early, and hence the importance of recognising these syndromes. Here, we review the neurological features of specific autoinflammatory syndromes and our approach (as adult neurologists) to their diagnosis.
- neuroimmunology
- immunology
- rheumatology
- clinical neurology
- haematology
Statistics from Altmetric.com
Introduction
Autoimmune diseases are characterised by a dysregulated adaptive immune system1 and are familiar foes to most neurologists. Autoantibodies or autoreactive T cells lead to a specific end-organ problems such as antibody-mediated postsynaptic dysfunction in myasthenia gravis or the central nervous system (CNS) involvement in anti-N-methyl-D-aspartate (NMDA)receptor encephalitis.2 3 In contrast, autoinflammatory syndromes, also called ‘periodic fever syndromes’, are caused by a dysregulated innate immune system and result in non-specific, and, therefore, widespread inflammation.4 Autoinflammatory syndromes may be inherited or acquired and can present in adulthood.5 Inherited (monogenic) forms include familial Mediterranean fever, tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS) and cryopyrin-associated periodic syndromes (CAPS). Acquired autoinflammatory syndromes include adult-onset Still’s disease, Schnitzler’s syndrome and more recently, vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome.6 7 Autoinflammatory syndromes are characterised by unexplained fever and systemic inflammation involving the skin, muscle, joints, serous membranes and eyes, along with elevated acute phase reactants.8 Neurological manifestations may be the presenting or predominant feature of the illness.9 Autoinflammatory syndromes present a significant diagnostic challenge to neurologists, because they are rare, the inflammation can affect the central or peripheral nervous systems and usually occur with non-neurological features. However, it is imperative to distinguish autoimmune diseases, infections and malignancy from autoinflammatory syndromes, because therapies are available to target the defective cytokine and alleviate further systemic or neurological damage.10 This review summarises the neurological aspects of the ‘common’ autoinflammatory syndromes and provides neurologists with a pragmatic approach when considering autoinflammation in the differential diagnosis.
Pathophysiology of autoinflammation
The innate immune system is the first line of defence against infection.5 There are evolutionarily conserved receptors (‘pattern recognition receptors’) within and on the surface of cells to detect infection and also trauma. Pathogen-associated molecular patterns are molecules, such as lipopolysaccharide, that are shared by large groups of microorganisms but are absent from the (human) host. During an infection, pathogen-associated molecular patterns bind to pattern recognition receptors, for example, toll- like or nod-like receptors, which activate a cascade of events including the release of the cytokines IL-1β and TNFα and lead to acute inflammation. A similar cascade of events is triggered when a cell is damaged through trauma; intracellular receptors detect damage-associated molecular patterns, such as DNA found outside the nucleus. Fever, vasodilation, endothelial adhesion, acute phase protein release from the liver (eg, C reactive protein and ferritin) and activation of other immune cells and complexes ensues. Aberrant activation of this innate immune response in the absence of infection or trauma leads to autoinflammatory syndromes (figure 1). The pathological abnormality in many autoinflammatory syndromes appears to be unregulated secretion of IL-1β and other proinflammatory cytokines. Monogenic autoinflammatory syndromes, such as familial Mediterranean fever, are caused by variants in single genes and follow a clear pattern of Mendelian inheritance.11 Acquired autoinflammatory syndromes, like Still’s disease, probably arise from combinations of common genetic variants together with other environmental factors. However, with the recent discovery of VEXAS, it may be that other poorly understood autoinflammatory syndromes result from single acquired mutations in somatic cells.7 In contrast to autoinflammation, autoimmunity results from aberrant adaptive immune responses (table 1). Consequently, autoimmune diseases have antigen-specific, organ-specific manifestations. However, if autoantigens are widespread, as is the case with systemic autoimmune disorders such as systemic lupus erythematosus, multiorgan involvement is common.
Autoimmunity versus autoinflammation
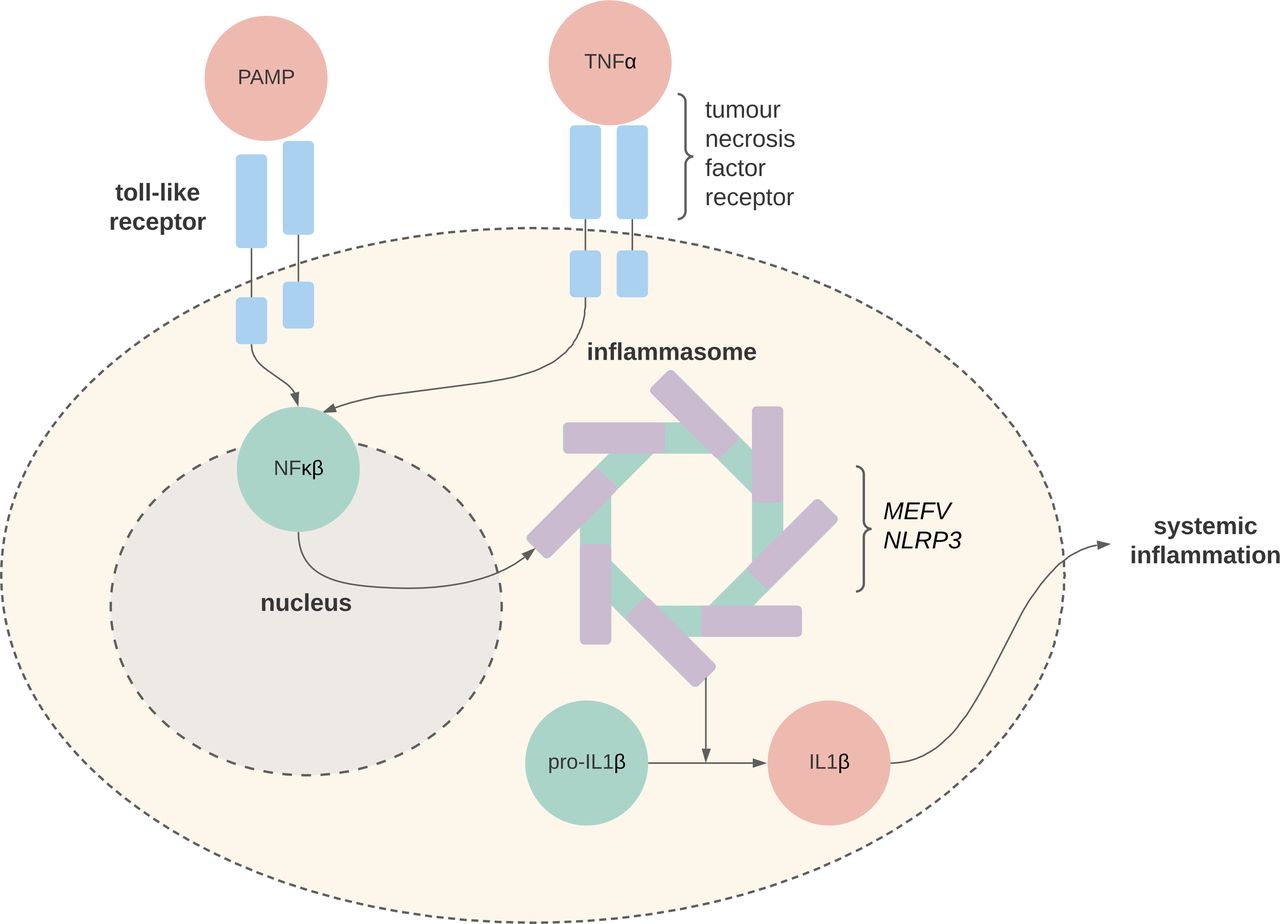
Simplified pathophysiology of autoinflammation. In the physiological state, pathogen-associated molecular patterns (PAMPs) bind to Toll-like receptors which lead to a cascade of intracellular signalling, including NFκβ-mediated activation of the inflammasome complex. In the pathophysiological state, genetic mutations in MEFV (familial Mediterranean fever) and NLRP3 (cryopyrin-associated periodic syndrome) lead to a constitutively activate inflammasome, unregulated production of IL1β and systemic inflammation. The pathophysiology of tumour necrosis factor (TNF) receptor-associated periodic syndrome is not well understood, but TNFRSF1A mutations lead to receptor misfolding and malfunction.
Clinical features
The non-specific nature of the innate inflammatory response means that virtually any organ system can be involved in autoinflammatory syndromes, including the central or peripheral nervous systems (figure 2).9 Recurrent fever unexplained by infection, malignancy or autoimmunity is the hallmark of autoinflammatory syndromes.11 The duration of the fever may vary from hours to weeks depending on the particular syndrome. Often patients will have been treated for presumed infection, without microbiological confirmation. Acute phase reactants are usually elevated during attacks. A maculopapular, urticarial or pustular rash is common. Arthralgia/arthritis and myalgia/myositis are common. Other typical features of autoinflammatory syndromes result from serosal inflammation, such abdominal pain from peritonitis, and chest pain or dyspnoea from pericarditis or pleuritis. Ocular inflammation can result in a range of ophthalmological features, including periorbital oedema, conjunctivitis and uveitis.12 Neurological involvement may occur in a range of autoinflammatory syndromes. Common manifestations include aseptic meningitis, sensorineural hearing loss, myositis and AA amyloid neuropathy. Less commonly reported neurological manifestations include brain and spinal cord inflammation, cerebrovascular disease and posterior reversible encephalopathy syndrome.9 13 14 Below, we outline the neurological features of some of the inherited (monogenic) and acquired autoinflammatory syndromes (table 2).
Summary of clinical and neurological features of selected autoinflammatory syndromes

Clinical features of autoinflammatory syndromes. SLE, systemic lupus erythematosus; SNHL, sensorineural hearing loss.
Inherited (monogenic) autoinflammatory syndromes
Familial Mediterranean fever
Familial Mediterranean fever is the most common monogenic autoinflammatory syndrome. It is an autosomal recessive disorder caused by mutations in the MEFV gene. MEFV encodes pyrin, which is part of the complex that detects pathogen-associated and damage-associated molecular patterns.15 Abnormal pyrin results in overexpression of IL-1β, which, in turn, leads to inappropriate neutrophil activation and bursts of systemic inflammation. Sephardic Jews, Arabs, Turks and Armenians are most affected. In 90% of patients, the onset is before 20 years of age. Common neurological manifestations include myalgia, often following exercise, and recurrent or chronic aseptic meningitis that responds to colchicine.16–19 CNS involvement may also occur.20 AA amyloidosis is a long-term complication and may result in peripheral neuropathy.14
TNF receptor-associated autoinflammatory syndrome
TRAPS is an autosomal dominant disorder caused by mutations in TNFRSF1A, leading to misfolding and malfunction of the TNF receptors.21 Its estimated prevalence is one per million. Ten per cent of patients present after the age of 30. Headache is common, but other neurological features are seldom reported.13 21 Small series of patients,9 and individual case reports,22 have described multiple sclerosis-like manifestations during typical TRAPS attacks.
Cryopyrin-associated periodic syndromes
CAPS is a group of autosomal dominant disorders caused by gain-of-function mutations in NLRP3, leading to a constitutively activated inflammasome.23 The estimated prevalence is 1–3 per million,24 and in one series, the median age of diagnosis was 31.5 years (0.5–75 years).25 Cold exposure may trigger disease relapses. Neurological features commonly include headache, sensorineural hearing loss and in severe cases, aseptic meningitis, papilloedema and seizures.23 26 Patients with low-penetrance NLRP3 mutations may develop severe CNS inflammation.9
Deficiency of ADA2
Deficiency of ADA2 (DADA2) is a polyarteritis nodosa-like vasculitis syndrome caused by mutations in ADA2, leading to an imbalance in differentiation of monocytes towards proinflammatory ‘M1’ macrophages.27 Onset in childhood is the rule, but adult-onset disease may occur. Clinical features resemble polyarteritis nodosa with unexplained fevers, livedo reticularis and multiorgan vasculitis. Haematological abnormalities are common. Half of the patients have neurological sequelae, most of whom have lacunar infarcts.28
Mevalonate kinase deficiency
Mevalonate kinase deficiency is caused by mutations in mutations in MVK.29 The spectrum of manifestations are determined by the degree of residual enzyme activity, including hyperimmunoglobulinaemia D with periodic fever syndrome and mevalonic aciduria.30 Neurologic manifestations are typical in mevalonic aciduria (developmental delay, cerebellar ataxia), but not hyperimmunoglobulinaemia D with periodic fever syndrome. However, mevalonate kinase deficiency almost always presents in the first year of life and is unlikely to pose a diagnostic conundrum for the adult neurologist.31 32
Acquired autoinflammatory syndromes
Adult-onset Still’s disease
Still’s disease is an autoinflammatory syndrome of unknown aetiology.33 The estimated prevalence is between 1 and 34 per million and it is a common cause of fever of unknown origin.34 It is characterised by fever, arthritis, evanescent rash and hyperferritinaemia. During attacks, the fever occurs once or two times per day and lasts less than 4 hours. Other features include splenomegaly, lymphadenopathy, abnormal liver enzymes and pleural or pericardial effusions. Severe disease may be complicated by macrophage activation syndrome, characterised by pancytopenia, hepatic dysfunction and coagulopathy. The diagnosis of Still’s disease still relies on the Yamaguchi criteria (table 3). A recent case series of 187 patients with Still’s disease found neurological involvement in 7.5% of patients.35 Aseptic meningitis was the most common manifestation, occurring in over half of patients with neurological involvement. The aseptic meningitis tends to be neutrophilic.36 Less common neurological manifestations included cranial neuropathies, encephalitis and ischaemic stroke. Those with neurological manifestations were more likely to have coexistent macrophage activation syndrome. Another series of patients with life-threatening Still’s disease described ‘neurological impairment’ in 25% of patients.37
Yamaguchi criteria for diagnosis of adult-onset Still’s disease49
Case 1
A 65-year-old man presented with low back pain, fevers and an erythematous rash on his chest. Acute phase reactants were elevated (C reactive protein >320 mg/L, neutrophils 17×109 /L) with thrombocytopenia (79×109 /L). Treatment for presumed pyelonephritis was started, although urine microbiology was negative. He then developed gait ataxia, left-sided ptosis with mydriasis and bilateral tinnitus and hearing loss. Cerebrospinal fluid (CSF) showed a white cell count of 9/µL (≤5) (74% lymphocytes), mildly elevated protein (0.65 g/L) and low glucose (2.2 mmol/L). Culture and viral/bacterial PCR of the CSF were negative. MR scan of brain showed patchy T2 hyperintensities in the cerebral white matter and contrast enhancement of the left oculomotor nerve. Audiometry confirmed bilateral sensorineural hearing loss. He was discharged home after some improvement in his symptoms, although the hearing loss persisted. One year later, he was seen in the hepatology clinic because of fluctuating liver enzymes with no cause found. One year later, he was hospitalised with left foot pain. He was febrile with elevated acute phase reactants (C reactive protein 186 mg/L, neutrophils 10.5×109 /L) and thrombocytopenia (25×109 /L). He then developed low back and shoulder pain, dyspnoea with patchy pulmonary infiltrates on chest radiograph, and atrial fibrillation. He was treated for presumed septic arthritis and/or pneumonia. The following day, he became rapidly comatose and was intubated and ventilated. CSF white cell count was 30 /µL (56% polymorphs). MR scan of brain showed widespread T2 white matter hyperintensities, most notably affecting the corpus callosum (figure 3). There were no neuronal antibodies in serum or CSF. CT scan of the chest, abdomen and pelvis found no underlying malignancy. Antineutrophil cytoplasmic antibody (ANCA), antinuclear antibody (ANA), extractable nuclear antigens (ENA), anti-double stranded DNA antibody (dsDNA), rheumatoid factor,
and antiphospholipid antibodies were negative. Six blood cultures were sterile. Serum ferritin was only mildly elevated (596 µg/L). Intravenous methylprednisolone (1 g/day for 5 days), intravenous immunoglobulins (2 g/kg), followed by oral prednisone (1 mg/kg/day), were administered with clinical and radiological improvement. Over the following 6 months, there were three unsuccessful attempts to reduce the prednisone dose below 20 mg/day, leading to a relapse with fever, rash (figure 4), arthralgia, encephalopathy, acute phase reactants and thrombocytopenia. An autoinflammatory genetic panel was negative (Invitae). He was diagnosed with Still’s disease according to the Yamaguchi criteria. Following the institution of maintenance prednisone (10 mg/day) and mycophenolate mofetil (2 g/day), he has had no further flares in 2 years, but has persistent mild cognitive impairment.

Widespread confluent T2 hyperintensities in cerebral white matter, including the corpus callosum. The changes resolved after intravenous methylprednisolone and immunoglobulins (not shown).

Urticaria-like erythematous rash associated with disease relapses during prednisone tapering.
Key message
The multisystem involvement of skin, joints, liver, lung, brain and cranial nerves in the context of fevers and elevated acute phase reactants suggests a systemic inflammatory process—infection, malignancy or autoimmunity. As workup for these was negative, autoinflammation was considered. The response to immunosuppression and relapse with prednisone tapering helped to confirm the diagnosis.
Schnitzler’s syndrome
Schnitzler’s syndrome is characterised by a monoclonal IgM gammopathy, chronic urticaria, fever, arthralgia and bone pain.38 Neurological complications are rare, but there have been isolated case reports describing sensorimotor neuropathy, encephalopathy, headache and vertigo.39–41 A case series of 281 patients reported peripheral neuropathy in 7% of patients.38
Case 2
A 68-year-old man was diagnosed with Schnitzler’s syndrome in 2012 after presenting with fevers, night sweats, chronic urticaria, elevated inflammatory markers, IgM kappa gammopathy and widespread lymphadenopathy on CT of his chest, abdomen and pelvis. His rash and laboratory measurements improved with colchicine and prednisone. In late 2019, he developed subacute onset of cognitive impairment, falls and ‘hiccups’ without improvement. In early 2020, he was admitted to hospital after a tonic-clonic seizure. Neurological examination showed confusion, gait ataxia and recurrent attacks of myoclonus with impaired awareness (online supplemental video 1). Acute phase reactants were elevated. CSF demonstrated mildly elevated protein (0.6 g/L) with a normal cell count. MR scan of brain showed generalised cerebral atrophy. Electroencephalogram showed frontal intermittent rhythmic delta activity. Work-up for infection, malignancy and autoimmunity was negative. An empiric trial of intravenous methylprednisolone (1 g/day for 5 days) followed by oral prednisone (1 mg/kg/day) resulted in complete resolution of the neurological symptoms and urticaria.
Supplementary video
Key message
In patients with known autoinflammatory syndromes, new neurological problems can be a manifestation of the disease and may improve with more aggressive immunosuppression.
Synovitis, acne, pustulosis, hyperostosis and osteitis
Synovitis, acne, pustulosis, hyperostosis and osteitis (SAPHO) syndrome includes a variety of osteoarticular disorders—synovitis, osteitis, hyperostosis, enthesitis (inflammation of entheses, where tendons or ligaments insert into bone)—associated with acne or pustulosis (large fluid-filled blister-like areas).42 Spinal lesions, including spondylodiscitis, are common, but rarely cause spinal cord compression.43 Hypertrophic pachymeningitis is well described in SAPHO.44–46
Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic
VEXAS is a recently described adult-onset autoinflammatory syndrome that occurs in men and is associated with a range of clinical phenotypes including relapsing polychondritis, Sweet’s syndrome, polyarteritis nodosa and giant cell arteritis.7 The disease is caused by acquired somatic mutations in the UBA1 gene.
Case 3
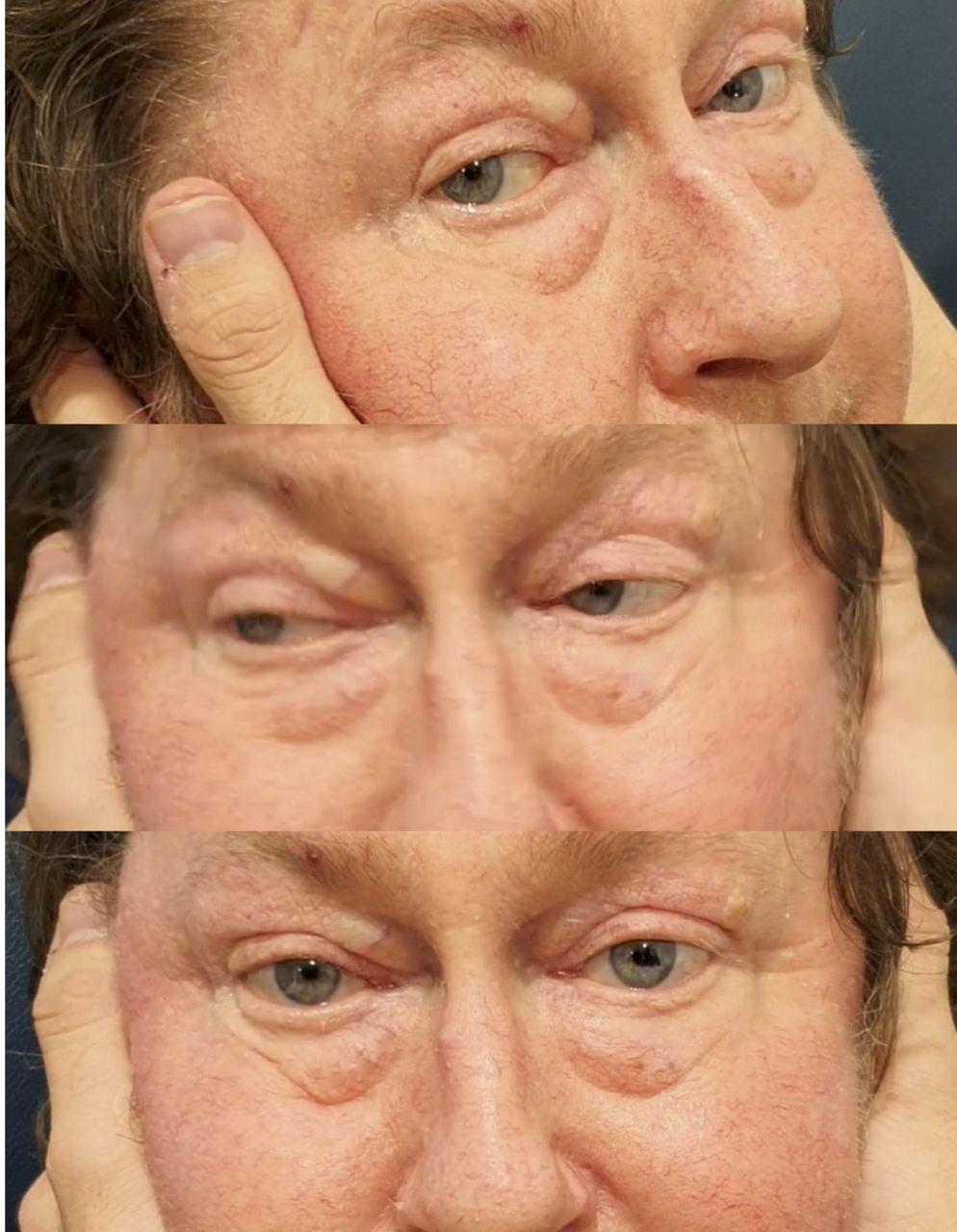
A 68-year-old man was diagnosed with possible Sweet’s syndrome in 2015 after presenting with relapsing episodes of fever and rash associated with an elevated C reactive protein (201 mg/L). Other features included a normocytic normochromic anaemia (haemoglobin 89 g/L, mean cell volume 97 fL), neutropenia (1.7×109 /L) and acute kidney injury (creatinine 178 mmol/L). Work-up for underlying infections, autoimmune disease and malignancy was unrevealing. Skin biopsies were reported as ‘papular urticaria’, ‘chronic urticaria’ and ‘leucocytoclastic vasculitis,’ although none were convincing for a neutrophilic dermatosis. He was initially trialled on topical corticosteroids and colchicine, but only responded well to oral prednisone >20 mg/day. Over the following 5 years, he presented with multiple disease flares, characterised by fevers, lethargy, rash, joint/muscle pain and an elevated C reactive protein. Disease flares triggered trials of different steroid-sparing agents, including methotrexate, azathioprine, cyclosporine and mycophenolate. In mid-2020, he presented with a 4-day history of severe right-sided retro-orbital headache. His temperature was 39.1°C. There was bilateral periorbital swelling and conjunctivitis (figure 5). C reactive protein was 197 mg/L. Serial blood cultures were negative. His dose of prednisone was 20 mg/day, and this was increased to 30 mg/day, along with antibiotics for possible periorbital cellulitis. Two days later, his headache evolved into right-sided retro-orbital pain, diplopia and sudden bilateral hearing loss. All movements of the left eye were severely restricted (figure 5). Hearing and the head impulse test were abnormal bilaterally. The remainder of the ocular and cranial nerve examination was unremarkable. MR scan of brain/orbits and CT cerebral venogram were normal. Audiogram confirmed moderate-to-severe bilateral sensorineural hearing loss. A temporal artery biopsy showed intimal lymphocytic inflammation without giant cells or elastic lamina fragmentation. Although the histology was not diagnostic of giant cell arteritis, the dose of prednisone was increased to 80 mg/day, with clinical improvement over a few days. The Blueprint Genetics Autoinflammatory Syndrome Panel did not show a pathogenic mutation. Following the description of VEXAS, a somatic UBA1 mutation was found. Ongoing treatment is with tocilizumab (162 mg/week), prednisone 20 mg/day and intravenous immunoglobulin (2 g/kg/month). His symptoms have been stable, but the vestibulopathy persists (figure 6). We hypothesise that the mechanism of ophthalmoplegia, sensorineural hearing loss and bilateral vestibulopathy was polycranial neuritis, given there was no evidence of orbital inflammation on brain imaging.

Restricted movements of the left eye, periorbital swelling and conjunctivitis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Positive head impulse test.
Key message
The underlying cause of many autoinflammatory syndromes remains elusive and ‘undifferentiated autoinflammatory syndrome’ is a common ‘final’ diagnosis. This patient had both Sweet’s-like and giant cell arteritis-like phenotypes, although the clinical features and histology were not typical for either. In patients with an undifferentiated autoinflammatory syndrome, treatment can be started empirically, but this should not prevent further diagnostic workup. Increasingly, genetic panels (eg, Blueprint Genetics or Invitae) are used to search for a specific diagnosis, but novel disease-causing mutations such as UBA1 (VEXAS) are not always included in these panels.
Haemophagocytic lymphohistiocytosis
This is a life-threatening syndrome caused by loss of down regulation of activated macrophages and lymphocytes.47 It can be thought of as a common endpoint of excessive immune activation, occurring in the context of infection, malignancy or autoinflammatory syndromes. When haemophagocytic lymphohistiocytosis-like features occur in Still’s disease, the term ‘macrophage activation syndrome’ is often used. Typical features of haemophagocytic lymphohistiocytosis are fever, high serum ferritin, hypertriglyceridaemia, hepatosplenomegaly with liver dysfunction, anaemia and thrombocytopenia. Most patients have haemophagocytosis on bone marrow biopsy. Neurological features include encephalopathy, seizures and posterior reversible encephalopathy syndrome.47
Diagnosing autoinflammatory syndromes
When to consider an autoinflammatory syndrome in adult neurology?
The diagnosis of autoinflammatory syndromes is challenging. From an adult neurologist’s perspective, autoinflammatory syndromes should be considered in a patient with a neurological disorder with:
Unexplained evidence of systemic inflammation (eg, fever, elevated acute phase reactants), especially when there are remitting and relapsing symptoms.
Unexplained multisystem disease (eg, rash, arthralgia, hepatic or cardiorespiratory involvement).
Improvement with corticosteroids.
The neurological manifestations of autoinflammatory syndromes are diverse, but certain disorders are more likely to be part of an autoinflammatory syndrome:
Aseptic meningitis or meningoencephalitis, particularly if neutrophilic.
Sensorineural hearing loss on a background of multisystem disease.
Peripheral neuropathy on a background of multisystem disease.
Diagnostic workup and management
The diagnostic workup of autoinflammatory syndromes from a neurologist’s perspective involves several steps. First, routine workup for presenting neurological problems is the most important step. Second, confirm that there is systemic inflammation, through documentation of fever and elevated acute phase reactants (C reactive protein, neutrophil count and ferritin). Elevation of plasma or serum cytokines (eg, IL-1β, IL-6, TNF) are transient and non-specific, and, therefore, not helpful. Third, exclude infection, malignancy and autoimmune disease as the underlying cause—this may entail blood cultures, viral and bacterial serology/PCR (eg, HIV, hepatitis, cytomegalovirus, Epstein-Barr virus, Bartonella sp), CSF culture with bacterial/viral PCR, autoimmune markers (eg, ANA/ENA, dsDNA, ANCA, rheumatoid factor), serum protein electrophoresis and imaging of the chest, abdomen and pelvis as is recommended in fever of unknown origin. Fourth, confirm that there is multisystem involvement: dermatological (eg, urticaria), haematological (eg, anaemia, thrombocytopenia), cardiorespiratory (eg, pleural or pericardial effusion), musculoskeletal (eg, tender joints) or hepatic (eg, abnormal liver enzymes) disease. Finally, if autoinflammation remains a likely cause, seek consultation with a clinical immunologist or a rheumatologist. When doing so, it is important to remember that many causes of inflammation of the central or peripheral nervous system are not associated with fever, elevated acute phase reactants and multisystem involvement. For example, both NMDA-receptor encephalitis and herpes simplex virus encephalitis can have fever, but neither disorder typically has evidence of inflammation outside of the CNS. Therefore, in the setting of CNS inflammation, elevated acute phase reactants and multisystem involvement, these disorders are less likely.
It is at this stage that an empirical trial of corticosteroids should be entertained. Pulsed intravenous methylprednisolone (500–1000 mg/day for 3–5 days) followed by oral prednisone (starting at 1 mg/kg/day, followed by a slow taper) is appropriate in undifferentiated autoinflammatory syndromes with significant neurological complications. Request an autoinflammatory genetic panel (eg, those offered by Blueprint Genetics or Invitae) in patients who relapse during the prednisone taper. Colchicine has anti-inflammatory properties based on the inhibition of leucocyte chemotaxis and is helpful in some autoinflammatory syndromes, such as familial Mediterranean fever. Other steroid-sparing agents are often required if symptoms relapse when the dose of prednisone is tapered. Biological agents, including IL-1 blockers (eg, anakinra, canakinumab), IL-6 blockers (eg, tocilizumab) and TNF blockers (eg, etanercept), are particularly effective in controlling many of the monogenic and acquired autoinflammatory syndromes.48
Concusions
Autoinflammatory syndromes are rare but increasingly recognised causes of systemic and neurological disease. Adult neurologists should remain vigilant that a patient presenting with unexplained neurological disease along with fever and elevated inflammatory markers may have an autoinflammatory syndrome, particularly if infection, malignancy and autoimmune disease have been excluded. Early consultation with rheumatology and/or immunology to assist with further investigation is essential, with discussion about the risks and benefits of empirical treatment with corticosteroids, colchicine or targeted immunomodulatory therapies.
Key points
Autoinflammatory syndromes result from dysregulated activation of the innate immune system.
They are characterised by recurrent fever, elevated acute phase reactants and multisystem inflammatory disease.
They have diverse neurological manifestations, but common presentations are aseptic meningitis, meningoencephalitis, sensorineural hearing loss and peripheral neuropathy.
Neurologists should consider autoinflammatory disease in patients with (1) unexplained fever and elevated acute phase reactants, especially when there is a remitting and relapsing course, (2) unexplained multisystem disease and (3) no evidence of infection, malignancy or autoimmune disease.
Further reading
Stojanov S, Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol 2005;17:586–99.
Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun 2018;93:24–36.
Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020;383:2628–38.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to thank Dr Lucy Lu, from the Department of Ophthalmology at Greenlane Clinical Centre, Auckland, New Zealand, for providing the images for case 3. We would also like to thank Dr Nikhar Shah, from the Department of Neurology at Auckland City Hospital, Auckland, New Zealand, for assisting with the filming of video 1 for case 2.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was published Online First. The abstract has been modified slightly.
Contributors WKD was commissioned to write the article. AJ provided immunology expertise. NEA provided neuroimmunology expertise. All authors contributed to writing and approving the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned. Externally peer reviewed by Neil Scolding, Bristol, UK.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Editors’ commentary
Other content recommended for you
- Familial Mediterranean fever (FMF) and beyond: a new horizon. Fourth International Congress on the Systemic Autoinflammatory Diseases held in Bethesda, USA, 6–10 November 2005
- Classification criteria for autoinflammatory recurrent fevers
- The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist
- Autoinflammatory diseases: update on classification diagnosis and management
- Visual loss with chronic meningeal and systemic inflammation
- Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study
- Cutaneous signs and mechanisms of inflammasomopathies
- Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers
- Inheritance of autoinflammatory diseases: shifting paradigms and nomenclature
- Systematic evaluation of nine monogenic autoinflammatory diseases reveals common and disease-specific correlations with allergy-associated features