Article Text

Abstract
When asked to assess patients in an intensive care unit (ICU) who have respiratory muscle weakness, oropharyngeal weakness and a vulnerable airway, our immediate thought may be of Guillain-Barré syndrome or myasthenia gravis, but there are many other possible causes. For example, previously unrecognised chronic neurological conditions may decompensate and require ICU admission. Clinicians can use various clinical clues to help recognise them and need to understand how patterns of weakness reflect differing causes of reduced consciousness on ICU. Additionally, patients admitted to ICU for any reason may develop weakness during their stay, the most likely cause being ICU-acquired weakness. Assessing patients in ICU is challenging, hampered by physical barriers (machines, tubes), medication barriers (sedatives) and cognitive barriers (delirium, difficulty communicating). Nonetheless, we need to reach a clinical diagnosis, organise appropriate tests and communicate clearly with both patients and ICU colleagues.
- intensive care
- myopathy
- neuropathy
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Statistics from Altmetric.com
Introduction
It is challenging to assess patients in intensive care unit (ICU),1 since they are often confused, sedated, intubated, in pain, distressed or potentially locked in.2 We should begin with a careful review of their notes, a discussion with staff who know the patient, and a telephone call to the family. The patient’s journey may have been long and complicated, perhaps beginning at another hospital (figures 1 and 2).

The phone is often more useful than our patellar hammer for intensive care unit neurological assessment.

Emphasise care not intensity.
Personal protective equipment, machinery, tubes, sedation and neuromuscular blockers can hamper our neurological assessment; patients are often so ill that arranging investigations is difficult.
A neurological assessment should ideally take place when sedation has been withdrawn. Drugs may take days to clear, despite claims to the contrary. After five half-lives, we can feel more confident that any remaining drug effect is negligible (eg, the half-life of fentanyl is 6 hours) but it can take longer, for example, with hypothermia or organ failure. Propofol is widely used in ICU, potentiating GABAA receptor activity; it usually wears off within minutes, but following prolonged or repeated administration, it can accumulate and linger in peripheral tissues.1
Neuromuscular junction blockers facilitate intubation and mechanical ventilation; their effects usually subside within a few hours of withdrawal. In patients receiving them for more than about a week—and particularly if they have renal failure, metabolic acidosis, hepatic disease, hypermagnesaemia or simultaneous corticosteroid or aminoglycoside exposure—prolonged neuromuscular blockade may develop, and last far longer than pharmacological measures alone might predict.3
Despite these difficulties, neurologists often find themselves assessing strength in the following groups of ICU patients:
Those with new-onset respiratory and/or oropharyngeal weakness, from various neurological disorders.
Chronic neuromuscular disorders, potentially previously unrecognised, that have decompensated.
Patients with reduced levels of consciousness. What do we learn from assessing them for weakness?
Patients admitted to ICU for any reason who become weak during their ICU admission. What are the causes of their weakness?
Box 1 highlights common causes for each of these categories.
The most common causes of weakness in intensive care unit (ICU)
Acute new-onset weakness requiring ICU admission
Neuropathy
Guillain-Barré syndrome
Neuromuscular disorders
Myasthenia gravis crisis
Muscle disorders
Electrolytes (often low serum potassium or phosphate)
Rhabdomyolysis
Spinal cord disorders
Hyperacute or acute myelopathies
Cord compression (epidural abscess, malignant, degenerative)
Trauma
Stroke both ischaemic and haemorrhagic. This can suddenly complicate bacterial and mycobacterial meningitis with cord infarction from vasculitis
Inflammatory, for example, infective, postinfective and neuromyelitis optica spectrum disorders
Disorders with acute-on-chronic decompensation
Motor neurone disease
Acid maltase deficiency
Myotonic dystrophy type 1—often presenting following surgery
Weakness developing after ICU admission
ICU-acquired weakness
Thresholds for test results (for example, guiding admission to or discharge from ICU) are particularly challenging, and require careful reflection and assessment as well as good communication. The first consideration should be how to approach such assessments and how to communicate with ICU colleagues about concerns that all share at such moments.
Assessment of patients with ventilatory and/or oropharyngeal weakness, and our interaction with ICU colleagues
Patients with neuromuscular respiratory weakness may superficially look comfortable. Thus, in those with rapidly evolving neuromuscular weakness, we must specifically look for signs of respiratory failure, clinically and whenever possible with bedside spirometry.
Box 2 highlights some important aspects to address on examination and the types of spirometry measurements that help identify respiratory muscle weakness.
Assessment of patients with potential respiratory or oropharyngeal weakness who may need intensive care unit (ICU) admission
Breathing
Ask the patient if they feel breathless (the increased respiratory medullary drive remains unsatisfied by afferent feedback because of muscle weakness)
Are they breathless when swallowing or speaking?
Look for increased respiratory rate—rapid shallow breaths
Check for tachycardia
Look for a sweaty brow
Assess strength of neck flexion: neck muscle strength mirrors that of the diaphragm (as observed in poliomyelitis) as they share the same segmental innervation
Assess accessory muscle use—for instance look and palpate the sternocleidomastoids, or look for nostril flaring with breathing
Observe the diaphragm. At rest, the diaphragm does most of the work: on inspiration it descends, moving the abdomen outwards and elevating the lower ribs. A weakened diaphragm might not be able to descend, giving ‘paradoxical’ abdominal movement in which the abdominal wall retracts on inspiration. This means the vital capacity has fallen to 10% of normal (about 500 mL in the average adult), dangerously close to the tidal volume of 400 mL
Speech and bulbar function. Impaired bulbar involvement leaves the airway unprotected with risk of aspiration, and limits clearance of secretions with risk of mechanical upper airway obstruction, particularly when supine. Clinical features include:
Increasingly nasal speech or speech that becomes dysarthric or quiet
Broken or staccato speech
A weak cough (important to listen to a voluntary cough)
Swallowing may be followed by coughing or indistinct ‘gurgly’ speech, indicating aspiration
Choking
Fluid regurgitation through the nose
Diminished palatal reflex
Portable spirometry—the ‘20/30/40’ rule that may often trigger ICU admission29 48 66 67
Forced vital capacity of<20 mL/kg (normal 60–70). A vital capacity of 30 mL/kg is associated with a weak cough and atelectasis. A fall in vital capacity of over 15%–20% from upright (sitting or standing) to supine reflects diaphragm weakness
Peak inspiratory mouth pressure of <30 cmH2O (normal>70)
Peak expiratory mouth pressure of <40 cmH2O (normal>100). This degree of weakness implies an insufficient cough to clear secretions adequately
Facial weakness can lead to inaccurate measurement in all these indices because of failure to make a good seal (with leakage), but an oro-nasal mask fitted over the mouth and nose and attached to the spirometer can reduce leakage. An alternative is to use the sniff nasal inspiratory pressure (normal>70 cmH2O). A tight, non-leaking plug is inserted into a nostril and connected to the spirometer; the patient inhales quickly and deeply through the unobstructed nostril
If no spirometry is available, then counting aloud to 20 in a single breath is reasonably reassuring (vital capacity >1.5 L); the clinician might count out aloud with the patient at a rate of 2 numbers per second
Some clinicians estimate the vital capacity by multiplying the number reached in a single breath by 100
The trend of measurements is important: a rapid decline even with normal values (and certainly a fall of more 30% from baseline) requires action
Clinicians must not rely on the ubiquitous peak expiratory flow metre; the rate of flow is proportional to the radius of a tube, and so peak expiratory flow rate provides a crude estimate of airway calibre, but not of respiratory muscle strength
ECG monitor, blood pressure and pulse oximetry for potential dysautonomia
Neuromuscular weakness impairs chest expansion and so reduces functional residual capacity, so the weakened diaphragm works harder since the lungs are less distensible at lower volumes. The impoverished lung expansion causes microatelectases and ventilation/perfusion mismatch with consequent hypoxaemia. The compensatory tachypnoea with small tidal volumes exacerbates atelectasis, further reducing lung compliance and increasing the mechanical load.
The clinical examination and spirometry measurements can identify significant respiratory muscle weakness long before hypoxaemia and even later hypercapnia develop. Hypercapnia presages impending catastrophic decompensation with respiratory arrest, so our assessment can be life-saving.4
We need to discuss with ICU colleagues any patient suspected of having conditions such as Guillain-Barré syndrome or myasthenia gravis. It is useful to assess patients jointly with ICU colleagues: we benefit from each other’s expertise and it helps establish an early rapport between the patient, the ICU team and ourselves. It is also useful to agree about thresholds that might trigger ICU admission, for instance spirometry results.
Patients with Guillain-Barré syndrome often deteriorate over a few days; reaching a nadir by about day 10. Thus, there may be time for repeated discussions but myasthenic crises are unpredictable. Early engagement with ICU must be the rule.
Remember to ask specifically for a respiratory muscle assessment in patients referred from outside hospitals by phone; it is easy to remain falsely reassured since patients superficially seem comfortable and their oxygen saturation remains normal.
Acute new-onset neuromuscular disorders with respiratory and/or oropharyngeal weakness, requiring ICU admission
Neuropathies
Guillain-Barré syndrome
This condition is heterogeneous and remains a clinical diagnosis.5 Common features include:
Early hip or lower back ‘strain’ (an orthopaedic consultation might begin the clinical journey).
Distal sensory symptoms without sensory signs.
Then a rapid symmetrical often proximal weakness, reflecting conduction block in nerve roots, usually legs before arms.
Deep tendon reflexes are frequently lost, although not invariably, and not infrequently just ankle jerks in the first few days: usually reflexes are lost by the time the limb cannot overcome gravity.
Bilateral facial weakness occurs in about half of patients.
Bulbar problems and other cranial nerve abnormalities develop in many patients.
One-third of patients are admitted to ICU because of respiratory insufficiency, oropharyngeal weakness, dysautonomia or some combination.
The severity of respiratory and limb weakness mirror each other (unlike in myasthenia) and potentially may be sufficiently profound to mimic brainstem death.6
Fasciculation and myokymia occur in early Guillain-Barré syndrome, manifesting as a brisk response to direct muscle percussion, potentially reflecting changes in axonal membrane potential, see figure 3.7–9
Repeating investigations can be valuable.
During week 1, cerebrospinal fluid (CSF) and a nerve conduction study are often almost normal, although sural sensory nerve sparing on nerve conduction is a useful early clue, peculiar to early Guillain-Barré syndrome.10
By week 2, CSF protein is raised in about 90% without cells (albuminocytological dissociation) and nerve conduction identifies demyelination in 90% of cases in Europe and North America.
If these tests remain normal, reconsider the diagnosis, see Box 3.
Red flags against a diagnosis Guillain-Barré syndrome
Severe respiratory weakness but mild limb weakness—(think myasthenia gravis)
Asymmetry of weakness, or weakness confined to the legs—(this is possible with regional variants but diagnose with caution)
Prominent early bladder or bowel problems—(think myelopathy)
No sensory symptoms—(think anterior horn cell disease, neuromuscular, low potassium)
Fever or raised inflammatory markers such as serum C reactive protein—(think infection such as Lyme disease or HIV, vasculitis or malignant infiltration)
Sensory level on the trunk—(think myelopathy)
Progression beyond 4 weeks, whereas most reach a nadir by week 2—(think chronic inflammatory demyelinating polyradiculoneuropathy, malignant infiltration of the nerve roots, IgG1-subclass pan-neurofascinopathy, diphtheria or myelopathy if no facial weakness)
Investigations: cerebrospinal fluid (CSF) pleocytosis: In Guillain-Barré syndrome, the CSF may contain up to 50 cells/µL—(but think HIV, Lyme disease or malignant infiltration in those with more than 10 cells/µL). Note that HIV-associated Guillain-Barré syndrome does not invariably have CSF pleocytosis. Normal nerve conduction after week 2—(think myelopathy or malignant root infiltration)

Still from video. Left leg fasciculation and myokymia in early Guillain-Barré syndrome, globally areflexic but brisk response to direct muscle percussion, often found in demyelinating neuropathies, perhaps deserving more study.
Porphyria
I recall a polished, persuasive neurologist explaining to a captivated although initially sceptical audience on ICU that a surgical patient—who had mysteriously become weak and had a seizure following an unremarkable laparotomy—had acute intermittent porphyria.
Acute hepatic porphyrias (most frequently acute intermittent porphyria):
Attacks often begin with.
Prodromal anxiety, confusion, insomnia, occasionally psychosis followed by.
Abdominal colicky pain, back pain and dysautonomia often with hyponatraemia.
Sympathomimetic features including posterior reversible encephalopathy syndrome.
Seizures (about 10% of attacks).
Attacks may evolve to a motor-predominant axonal neuropathy (10%–40% attacks):
Beginning in the arms as frequently as legs, sometimes proximally and asymmetrically. Cranial nerves and respiratory muscles can become involved.
Myalgia is often prominent.
Reflexes may be retained, sometimes just ankle jerks are retained.
Pain or paraesthesia proximally (swimming trunks and onto torso) or distally.
Blisters in sun-exposed areas occur in variegate or hereditary porphyria.
Porphyrinogenic drugs (including alcohol), dieting, hormonal influences or sun exposure can precipitate attacks.
The first diagnostic step is to measure δ-aminolevulinic acid, creatinine and porphobilinogen (four times upper limit normal is diagnostic) in a light-protected urine sample.11
Diphtheria
In the early 1990s, several cases of diphtheria appeared among people returning from Russia. One such person in an ICU with a demyelinating neuropathy and raised CSF protein was being treated for Guillain-Barré syndrome; the penny dropped only when it emerged that someone else from the same trip had fallen ill with an almost identical history.
Corynebacterium diphtheriae causes a sore throat with considerable neck swelling (bull neck). ‘Diphtheria’ is Greek for leather, reflecting the leathery pseudomembrane covering the tonsils.
An exotoxin is produced that inhibits synthesis of myelin basic protein leading to demyelination, initially locally causing bulbar symptoms and then after haematogenous spread affects the limbs, a biphasic natural history.12 Features include:
Bulbar weakness 2 weeks after the sore throat, with dysphagia and nasal speech.
Pupillary constriction failure to accommodation (giving blurred vision) develops shortly afterwards.
Bulbar weakness might begin to improve but over the following 5 weeks or more limb weakness and distal sensory loss develop.
Cardiac involvement (myocarditis), with ECG showing ventricular ectopics or conduction abnormality (vagus denervation may also cause a tachycardia).
Demyelinating neuropathy identified on neurophysiology.
CSF results are variable, sometimes a pleocytosis but often albuminocytological dissociation.
Unlike Guillain-Barré syndrome, the slow biphasic onset mirrors a slow recovery time. In Guillain-Barré syndrome, the monophasic onset is rapid, often reaching a nadir by day 10, followed by a slow recovery.
In diphtheria, the facial nerve is less frequently affected than in Guillain-Barré.13
Antitoxin treatment offers little benefit once the neuropathy has begun.
IgG1-subclass pan-neurofascinopathy
This is the other neuropathy that rapidly culminates in respiratory failure. It resembles Guillain-Barré but responds to rituximab, though not to intravenous immunoglobulin. I have a low threshold for testing for nodal/paranodal antibodies in severe Guillain-Barré-like presentations.14 15
Other neuropathies
We are all likely to encounter patients with phrenic nerve palsies. The causes include:
Cardiac surgery.
Idiopathic—potentially a variant of brachial neuritis.
Malignant invasion.
Rarely with chronic inflammatory demyelinating polyradiculoneuropathy, a treatable cause.
Paradoxical diaphragm movement is a late clinical sign, with inward movement of the abdomen on inspiration when the patient is supine. X-ray of the chest may identify elevation of diaphragms. M mode (‘motion’) ultrasonography examines diaphragm movement, helping to confirm the diagnosis.16
Examples of other neuropathies that may require ICU admission include: toxins (eg, acute arsenic intoxication causing a demyelinating neuropathy, gastrointestinal symptoms and bone marrow suppression), drugs (eg, immune checkpoint inhibitors), beriberi, lymphoma neuropathies, Lyme disease, vasculitis, HIV-associated neuropathies and rabies.4
Neuromuscular disorders
Myasthenia gravis (MG)
Myasthenic crisis affects about 10% of people with myasthenia, usually within the first 2 years of diagnosis, defined as respiratory failure secondary to respiratory or upper airway muscle weakness or both.
The crisis may be provoked by:
Respiratory infection.
Surgery.
Electrolyte changes.
Thymoma recurrence.
Pregnancy.
Inadequate myasthenic treatment or inappropriate treatment with drugs that exacerbate myasthenia (notably sedatives and some antibiotics).
In about a third, no cause is found.
Myasthenic crisis is unpredictable and extubation fails in about a quarter of cases, so clinicians must take particular care when patients leave ICU. Two-thirds of deaths occur after ICU discharge, similar to the figures for Guillain-Barré.17 18 Box 2 outlines the clinical observations for patients before ICU admission: these will need to be reinstated.
There is a role for non-invasive ventilation in a myasthenic crisis, initiated by spirometry criteria or respiratory muscle fatigue or blood gas results. (Non-invasive ventilation would not be appropriate for Guillain-Barré since respiratory muscle failure in this situation, unlike in myasthenia, is followed by inexorable decline with little chance of reversibility). In myasthenia it may mean avoiding, shortening the duration of intubation or preventing the need for reintubation.19–21
Major treatments for myasthenic crisis include:
Intravenous immunoglobulin or plasma exchange.
High-dose prednisone (60–100 mg daily) initiated concurrently; the corticosteroid begins to work after 2 weeks, just as the effects of intravenous immunoglobulin or plasma exchange start to wane.
Acetylcholinesterase inhibitors are generally stopped, to avoid excessive bronchial secretions.
Muscle-specific kinase antibody myasthenia has a high frequency of respiratory crises and differs from acetylcholine-receptor antibody-positive myasthenia in several ways. A recent patient taught me a great deal about this condition (see Box 4).
Case history
A woman in her 80s presented with new and severe bulbar palsy, ptosis, facial weakness, normal eye movements, ‘triple furrowed’ tongue without fasciculation, weak neck extensors (‘head drop’) and type 2 respiratory failure, but with strong limbs, reflexes present and plantar responses down.
Clinically, this looked like myasthenia gravis.
She started non-invasive ventilation with bi-level positive airway pressure on intensive care unit (ICU) that sustained her throughout her ICU stay.
A limited nerve conduction (patient choice) showed no decrement on distal repetitive nerve stimulation.
She received corticosteroids, intravenous immunoglobulin and low-dose pyridostigmine (total 90 mg/day). There was no improvement and a week after starting low-dose pyridostigmine she became bradycardic with dramatic worsening of secretions.
We suspected antimuscle-specific tyrosine kinase myasthenia gravis, subsequently confirmed with positive muscle-specific tyrosine kinase antibodies. This condition:
Affects 5%–10% of people with generalised myasthenia.
Often has an acute rapid bulbar onset with respiratory crisis.
Tongue atrophy (triple furrowed).
Neck extensors weaker than neck flexors; acetylcholine receptor antibody-positive myasthenia usually causes weaker neck flexors.
Less frequently causes a decrement with distal repetitive nerve stimulation compared with acetylcholine receptor antibody-positive myasthenia.
Often a poor response to acetylcholinesterase inhibitors and conventional immunosuppression with prominent cholinergic side effects at low doses of pyridostigmine.
After stopping pyridostigmine, she started to improve with 3,4-diaminopyridine and oral salbutamol.68–70
Plasma exchange and rituximab are frequently helpful in this situation but she was frequently haemodynamically unstable with recurrent pseudomembranous colitis and felt therefore to be too ill to have these treatments.
She sang, ‘Myfanwy’ leaving ICU and has remained well ever since.
Botulism
Neurologists occasionally see botulism particularly among drug abusers. Clostridium botulinum spores contaminate muscle or subcutaneous injection sites, germinate and release neurotoxin that prevents acetylcholine release, at neuromuscular junctions and autonomic ganglia.
Ptosis, ophthalmoplegia with blurred vision, dysarthria, dysphagia and facial weakness develop. Poor pupillary reaction and dry mouth are common and three-quarters require mechanical ventilation.22
A neurologist suspecting botulism should start treatment. Neurophysiology will help identify a preganglionic neuromuscular junction problem but isolation of C. botulinum from pus or detection of toxin in serum are insensitive.6
Lambert-Eaton myasthenic syndrome
Lambert-Eaton myasthenic syndrome presents with:
Fluctuating proximal weakness (about three-quarters present with proximal leg weakness).
Reduced or absent deep tendon reflexes that may temporarily return, along with muscle strength following muscle contraction (postexercise facilitation).
Cholinergic dysautonomia with constipation, dry mouth or pupillary abnormalities.
Lambert-Eaton myasthenic syndrome may present with respiratory failure, an under recognised presentation in a condition that is already notoriously easily overlooked even in a calm neurology or neurophysiology clinic.23 If it presents with respiratory failure then there will also be generalised weakness, unlike the usual case with MG. Most have autoantibodies against the P/Q-type voltage-gated calcium channels.
Anterior horn cell diseases
Motor neurone disease may present with acute-on-chronic decompensation (see below).
Viruses including non-polio enteroviruses (D68 and 71) and West Nile virus can involve anterior horn cells, causing segmental, asymmetric weakness potentially involving respiratory muscles. Throat swabs for enterovirus PCR and anti-West Nile virus IgM in CSF confirm the diagnosis.24 25
Muscle
Neurologists will all see the following conditions that potentially require ICU admission:
Electrolytes disorders
Invariably tested in the emergency department but often unchecked. Look up the results—notably looking for hypokalaemia and hyperkalaemia, hypophosphataemia and hypermagnesaemia.26
Hypokalaemia from any cause can produce a flaccid, areflexic weakness and potentially respiratory muscle weakness.
Hypophosphataemia can follow hyperalimentation, particularly after periods of starvation before or during ICU admission and may also cause weakness.
Familial hypokalaemic periodic paralysis and thyrotoxic hypokalaemic periodic paralysis can rarely cause respiratory muscle weakness27; a normal acid–base balance distinguishes them from other causes of hypokalaemia.28
Anderson-Tawil syndrome can present with hypokalaemic paralysis; look for dysmorphism and associated cardiac conduction defects.29
Rhabdomyolysis may occur, for example, in:
The medication history needs to be scrutinised.
Chronic renal failure patients are at particular risk with colchicine and many have gout. I recall a patient with kidney disease taking colchicine who developed a neuromyopathy that involved the respiratory muscles. Amiodarone, vincristine and hydroxychloroquine are also important to consider.
Propofol infusion syndrome is rare and develops within days of administration, causing metabolic acidosis, cardiac dysfunction, renal failure and rhabdomyolysis. Patients with mitochondrial disease are particularly susceptible; neurologists should avoid using propofol in such patients.32 33
Idiopathic inflammatory myopathies
Patients with these disorders rarely present with respiratory muscle weakness, as may those with anti-Jo1-positive antisynthetase antibody and necrotising autoimmune myopathies.34
Spinal cord lesions or brainstem stroke mimicking Guillain-Barré syndrome
Severe spinal injury from C3 to C5 may result in a period of spinal shock, lasting up to 6 weeks, and characterised by a flaccid tetraparesis with all spinal segmental reflexes suppressed (including deep tendon reflexes) below the level of the lesion. A necrotising myelitis may similarly cause loss of deep tendon reflexes following extensive destruction of the spinal cord grey matter. The respiratory muscles are paralysed just like the limb muscles and so this clinical picture can resemble Guillain-Barré.
HyperCKaemia develops in some people with neuromyelitis optica spectrum disorders.35 Clinicians should have a low threshold for imaging the spine and should not feel dismayed after having imaged patients who ultimately have a lower motor neurone disorder (figure 4).36

Sagittal T2 MR cervical (left) and thoracic cord (right); increased signal and swelling cord from C3 down.
Box 5, summarises some clinical clues to distinguish myelopathy, Guillain-Barré syndrome or brainstem infarction from basilar artery thrombosis. Basilar artery thrombosis, particularly a mid-basilar stroke with bilateral pontine base ischaemia, sometimes involves the pontine tegmentum (as opposed to a ‘top of the basilar’ stroke), which may also cause a flaccid tetraparesis with ocular and bulbar findings, mimicking the Guillain-Barré spectrum with ophthalmoparesis.
Clues to help distinguish Guillain-Barré syndrome from acute myelopathy
Favouring Guillain-Barré syndrome
Facial weakness—found in half, both sides of the face simultaneously becoming weak or doing so sequentially
Distal paraesthesia, with sensory symptoms in the fingers by the time these reach the mid-calves
Sensory symptoms precede the sensory signs; sensory loss is often confined to distal impairment of vibration sense
Limb weakness is worse in the legs than the arms early on
Respiratory involvement is expected when the limbs cannot overcome gravity—respiratory muscle weakness mirrors limb weakness in Guillain-Barré syndrome but remember this actively needs to be sought (see box 2)
Autonomic dysfunction—about two-thirds have dysautonomia, with labile blood pressure, tachycardia or tachyarrhythmias more common than bradycardia. Note that excess vagus nerve activity (‘vagal spells’) with bradycardia and asystole may be provoked by stimulation (for example by tracheal suction) or spontaneously.
Favouring myelopathy
Marked sensory loss proximal to the hands or feet, or on to the trunk early-on
Sensory level on the trunk
Radicular pain in the arms (radicular leg pain may develop in Guillain-Barré syndrome)
Prominent sphincter disturbance
Weakness in the arms and legs is uniformly severe, or weakness is confined to the legs, or weakness is asymmetrical
Interruption to the sympathetic nervous supply may result early on in hypotension, bradycardia and hypothermia (unexplained hypotension and bradycardia in an unconscious intensive care unit patient should prompt consideration of a myelopathy)
Impalpable lower limb pulses may reflect spinal stroke secondary to, for example, aortic dissection
Favouring mid-basilar pontine stroke over ophthalmoplegic forms of Guillain-Barré syndrome
Very abrupt onset
Asymmetric limb weakness or unilateral onset of weakness; often there is a clear hemiparesis but with some additional abnormal signs contralaterally, such as brisk reflexes or an extensor plantar response
Crossed signs, for example, right facial weakness with left limb weakness (see figure 8)
Brisk deep tendon limb reflexes (occasionally deep tendon reflexes are initially diminished)
Pseudobulbar palsy—brisk facial reflex and jaw jerk (interruption of corticofugal fibres in the basis pontis destined for cranial nerves)
Eye signs are often asymmetric, whereas in Guillain-Barré syndrome the bilateral external ophthalmoplegia is usually symmetric. Common types of eye abnormalities include:
Unilateral conjugate gaze paresis (pontine lateral gaze centre dysfunction)
Unilateral internuclear ophthalmoplegia (medial longitudinal fasciculus dysfunction)
Nystagmus (types of central vestibular nystagmus)
Ocular skew
Ocular bobbing
‘Pontine pupils’ are pinpoint but still react to light
Bilateral damage of the medial pontine tegmentum results in stupor or coma. However, the collateral supply is often adequate, sparing the reticular activating system; sensation remains unaffected since the dorsolateral tegmentum is also often spared.
Many patients therefore remain awake and aware but with loss of motor function (locked-in); eye blinks or vertical eye movements show the patient is alert and intellectually preserved.
Frequently these patients report preceding (over days or weeks) transient attacks of diplopia, dizziness, bilateral leg or alternating limb weakness
Occasional patients report odd preceding symptoms, including auditory hallucinations (ischaemia of the auditory pathways), or spontaneous limb movements or posturing, mimicking a seizure.
Acute-on-chronic weakness, potentially unrecognised that decompensates requiring admission to ICU
The diaphragm is responsible for 70% of ventilation at rest and is our only working ‘bellows’ during rapid eye movement (REM) sleep, when there is both hypotonia of accessory respiratory muscles and reduced respiratory drive. An early feature of chronic respiratory muscle weakness, therefore, is oxygen desaturation and hypercarbia during REM sleep.37 The resulting sleep fragmentation and carbon dioxide retention can cause:
Nightmares
Morning headache
Grogginess and daytime somnolence
Delirium occasionally
Papilloedema in extreme cases
Signs of right heart failure if chronically hypoxic
Orthopnoea and dyspnoea on immersion also reflect diaphragm weakness, when the abdominal contents splint the weakened diaphragm.
Patients with neuromuscular weakness often do not have exertional dyspnoea, since their mobility is frequently impaired; however, if the limbs remain strong then exertional dyspnoea can be the presenting complaint, alongside the other symptoms of respiratory muscle weakness outlined above.
Functional residual capacity declines with neuromuscular weakness, so the weakened diaphragm works harder, with the lungs being less distensible at lower volumes. A trivial additional burden can lead to decompensation, maybe as trivial as constipation or an increased respiratory rate accompanying a mild fever.37
Table 1 summarises neuromuscular conditions that may present for the first time with significant respiratory weakness but with relatively preserved limb strength, and some clues to help identify them. Two conditions to highlight particularly are:
Motor neurone disease can present with diaphragm weakness (about 3% of patients) when there is only mild weakness elsewhere. These patients often have a brief preceding history (few weeks or months) of the respiratory symptoms outlined above. Examination usually identifies the tell-tale combination of upper and lower motor neurone signs with widespread fasciculation.38 In such patients, it is sometimes possible to establish non-invasive ventilation, but not always. Invasive ventilation prolongs survival, but is fraught with difficulty, not least from the profound loss of quality of life. It is particularly helpful to involve palliative care to help guide the decision-making process in this most challenging situation. Almost invariably, there is a decision to withdraw invasive ventilation, using judicious and adequate sedation for comfort and dignity.
Myotonic dystrophy type 1 patients often have respiratory and oropharyngeal muscle weakness with increased sensitivity to various respiratory depressants, including opiates. Postoperative complications are common; about 1 in 10 surgeries with general anaesthesia are complicated by respiratory problems, including prolonged respiratory depression, particularly following cholecystectomy.39 40
Conditions with acute-on-chronic compromise potentially presenting to ICU with no previous diagnosis
Table 2 summarises other neuromuscular conditions that generally will have been recognised long before respiratory muscle weakness precipitates a hospital admission; the declining respiratory muscle strength reflects disease progression. These patients will usually have discussed their likely natural history with neurologists, respiratory physicians and intensivists, including advance care planning and their preferences regarding their medical care and ceiling of care.
Conditions already diagnosed with worsening respiratory muscle weakness as part of their natural history
Assessing strength in patients with reduced levels of consciousness in ICU
Neurologists frequently see patients in ICU who have reduced levels of consciousness, trying to establish both the cause and prognosis. Motor examination is one aspect of the assessment.
Diffuse metabolic conditions commonly lead to patients having paratonia (oppositional resistance to passive movement), extensor plantars and myoclonus, but some also have focal weakness. Hemiplegia may be a feature of hypoglycaemia, uraemia, hyponatraemia or hepatic encephalopathy (also hepatic myelopathy).
Toxic conditions causing coma sometimes gives focal weakness. For example, ethylene glycol poisoning—characterised by a high anion gap acidosis with increased osmolar gap but without a distinctive breath odour—may be complicated by bilateral facial weakness and rarely a flaccid areflexic paralysis (with raised CSF protein, sometimes a pleocytosis); respiratory muscle weakness may begin a week after intoxication.41 42
Supratentorial mass lesion
Hemiplegia may be ipsilateral, a false localising sign reflecting ipsilateral uncal herniation that presses the contralateral cerebral peduncle against the tentorium cerebelli, distorting the contralateral corticospinal tract.
A large ipsilateral fixed pupil from pressure on the oculomotor nerve is, by contrast, a good lateralising sign.43
Intrinsic structural brainstem disease, see figures 5–7.
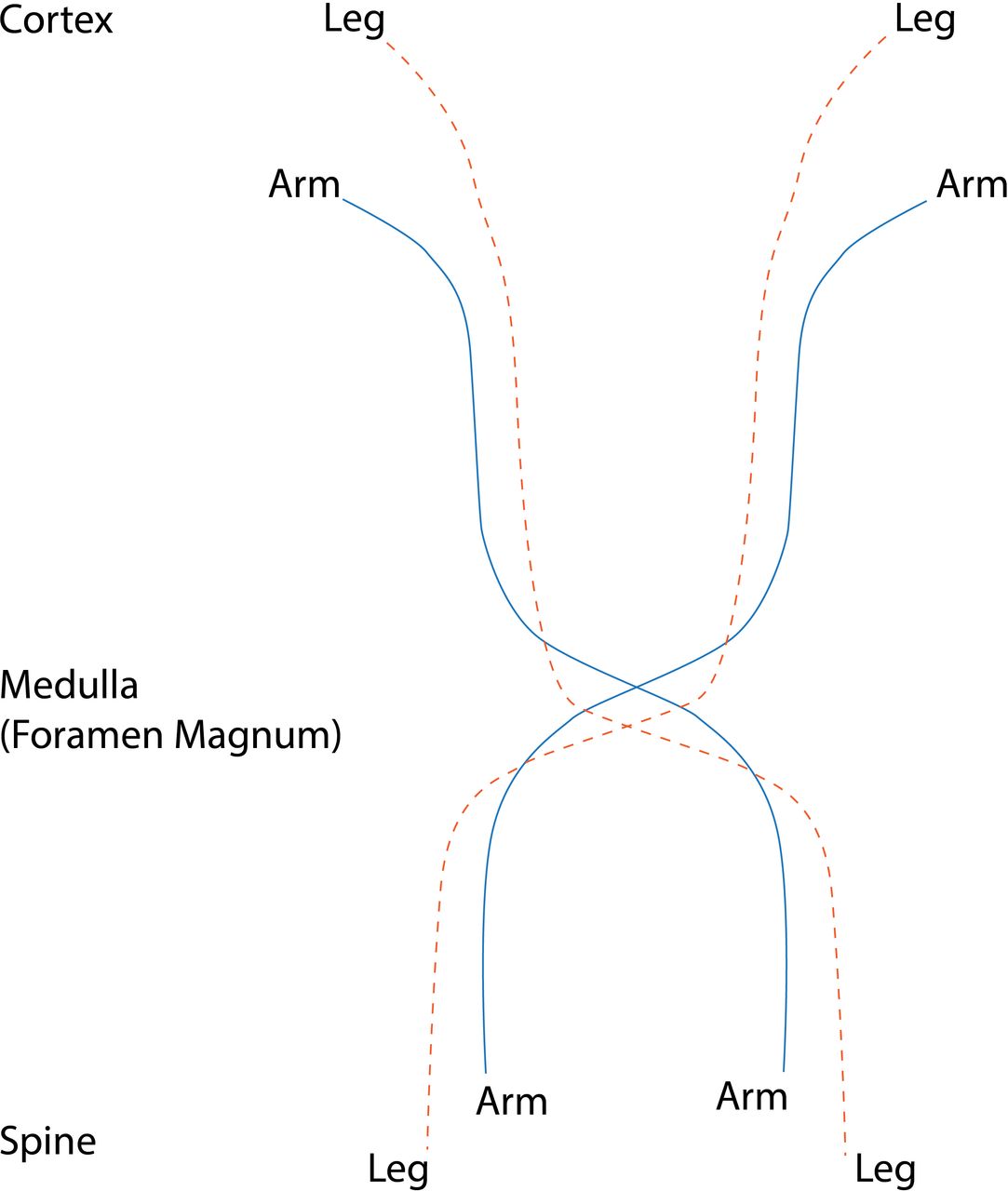
Corticospinal fibres descend in the cerebral peduncles and ventral pons before decussating in the medullary pyramid. Limb weakness is often bilateral but asymmetric; unusual patterns of weakness are possible, see figure 8.
Cranial nerves 3 and 4 (midbrain), 5, 6, 7 and 8 (pons) and 9, 10, 11 and 12 (medulla) potentially cause diplopia, facial or jaw weakness as well as dysarthria, dysphonia, dysphagia and tongue weakness.44 45

Basilar stroke. Hyperdense basilar artery on unenhanced CT of the brain. Diplopia, dizziness, leg weakness or alternating hemiparesis is common in the weeks before basilar stroke. Auditory hallucinations or seizure-like movements sometimes occur.75

MOG antibody disease. Signal change in superior cerebellar peduncles (axial FLAIR MR). Headache, drowsiness, gaze palsy, tetraparesis, ataxia, extensor plantars drowsy requiring intubation. Treated for Listeria and given intravenous methylprednisolone with steroid taper. By day 8, extubated, recovered well.

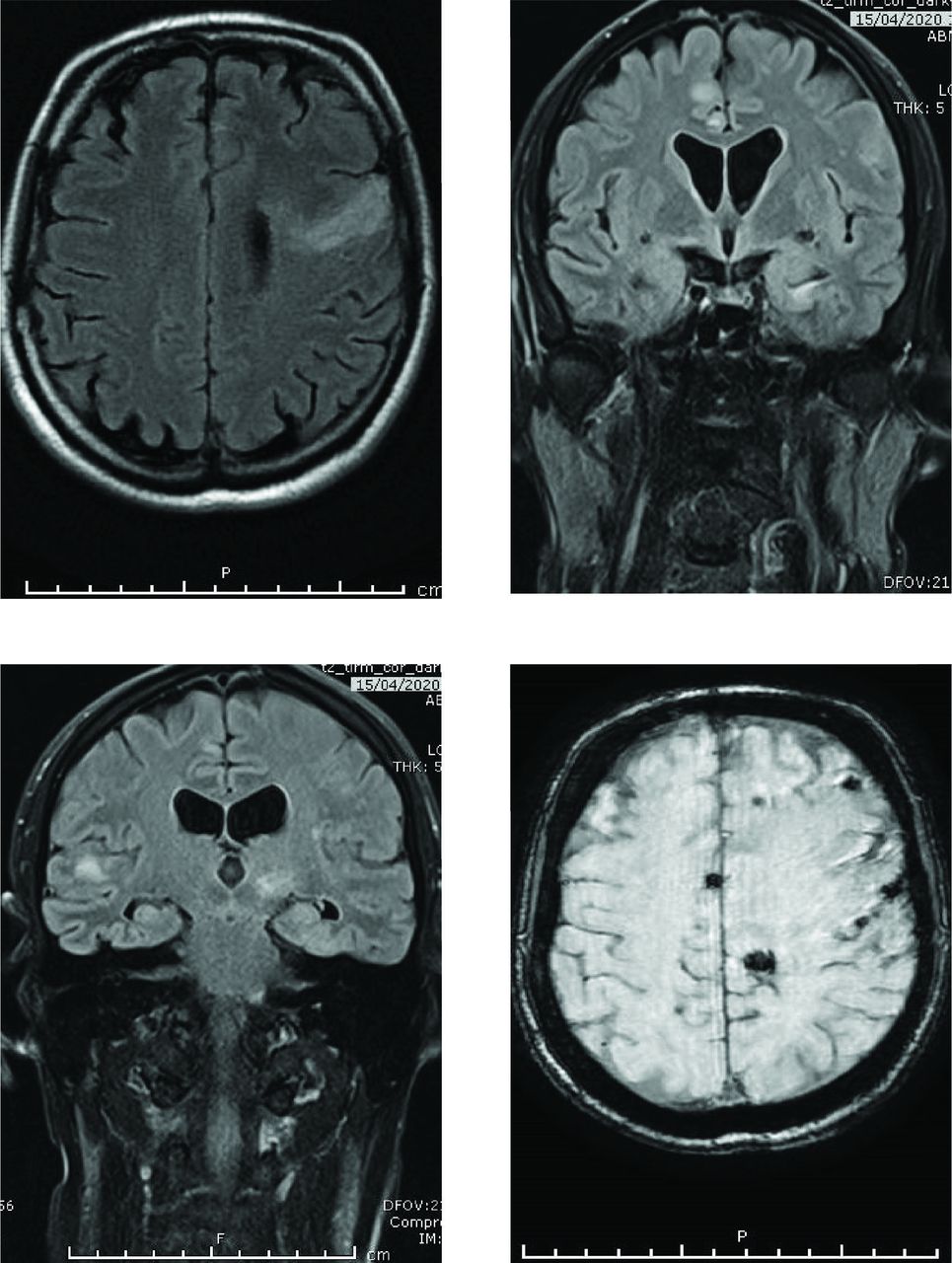
Varicella zoster virus (VZV) encephalitis. MRI axial FLAIR (top left) and coronal FLAIR (top right, bottom left) show lesions at grey, white and grey-white matter junction (therefore, may resemble emboli or metastases), both ischaemic and haemorrhagic, susceptibility-weighted imaging axial MR (bottom right). This patient had presented with a thoracic myelopathy and paraparesis. Cerebrospinal fluid (CSF) showed positive PCR for VZV DNA; but negative for other neurotropic viruses. Three weeks later the patient developed a tetraparesis, slow saccades, internuclear ophthamolplegia, ataxia, increasing drowsiness and irregular gasping respiration. He was not immunocompromised, and had no history of shingles. Persistently positive CSF PCR VZV DNA, and in ICU for 3 months. Intravenous acyclovir given for 2 months, prednisolone 60 mg for 5 days without taper. VZV myelopathy can be protracted and may be complicated by a VZV encephalitis; vasculopathy is a major pathogenic process in VZV encephalitis caused by viral infection of cerebral arteries.76 He still has bladder symptoms, left hemiparesis and frontal lobe cognitive problems but continues to recover. FLAIR, fluid attenuated inversion recovery.

Motor pathways descending to cross in the medulla. Leg fibres cross below arm fibres; a lesion may therefore cause weakness in both arms or a leg and opposite arm.
Patients who develop weakness while on ICU
Osler described wasting and weakness complicating prolonged sepsis. The term ICU-acquired weakness is modern parlance for Osler’s perceptive original description.46
ICU-acquired weakness encompasses
Critical illness myopathy, causing proximal symmetrical weakness, is two times as common as other forms ICU-acquired weakness and has a better prognosis.47
Critical illness polyneuropathy causes a flaccid, symmetrical predominately distal weakness with distal sensory loss.
Critical illness polyneuromyopathy is the term describing the many cases with both critical illness polyneuropathy and critical illness myopathy.
The respiratory muscles are involved in each, but the facial and extraocular muscles are generally spared.29 48 In this setting, extraocular muscle weakness raises suspicion of prolonged neuromuscular blockade; neurophysiology is helpful to test that clinical hypothesis, see table 3.
Nerve conduction studies in intensive care unit-acquired weakness
Incidence of ICU-acquired weakness
The incidence depends on the definition, and differs if defined by clinical or electrophysiological criteria, and by the population being considered. The important message is that it is common, affecting about half of patients after 1 week in ICU and two-thirds of people after 10 days who have sepsis.49 Many authors add together the MRC muscle strength from 12 muscles; a score below 48/60 defines ICU-acquired weakness but adds little to everyday practice.
Why are patients with ICU-acquired weakness referred to neurology?
The onset of weakness in ICU patients is often masked by severe underlying systemic illness, sedative medication and encephalopathy. Weakness may first come to attention when there is a difficulty weaning a patient from mechanical ventilation. Such difficulty implies a failure to pass a 30 min spontaneous breathing trial or reintubation within 48 hours.50
There are several potential causes, including weakened respiratory muscles from ICU-acquired weakness. Other causes include:
Airway and lung dysfunction, for example, relating to increased upper or small airway resistance.
Brain dysfunction, for example, relating to delirium or sedatives.
Cardiac dysfunction, for example, transitioning from mechanical ventilation to spontaneous breathing may affect the intrathoracic pressure and therefore ventricular preload and afterload.
Other medical problems, may including adrenal insufficiency, dysthyroid, electrolyte disorders such as low phosphate.
At other times, ICU staff may misinterpret reduced limb movements and ask a neurologist why the patient remains in coma. However, examination might establish that the patient can open their eyes, blink and move their eyes, signalling they are awake and aware, even though many have a delirium.
Risk factors for ICU-acquired weakness
These include:
Persistent systemic inflammatory response syndrome, an exaggerated responaw to various insults, including infection, trauma and surgery. The dysregulated release of acute-phase reactants and cytokines cause end organ damage. Sepsis is a particularly significant risk and sepsis-associated encephalopathy commonly accompanies ICU-acquired weakness, complicating its assessment.1 49
Muscle inactivity.
Hyperglycaemia.
Probably corticosteroids and neuromuscular blockers.
Possibly low serum albumin, parenteral nutrition and hyperosmolarity.
The pathophysiology of ICU-acquired weakness
There are likely to be many mechanisms, including:
Microcirculation dysfunction, endothelial activation secondary to systemic inflammatory response syndrome leading to microvascular leak, oedema, inflammation and hypoxia.
‘Battery failure’ (bioenergetic failure), with compromised mitochondrial oxidative phosphorylation secondary to, for example, inflammation, high plasma glucose and oxidative stress in critically ill patients. With insufficient ATP to fuel all metabolic pathways, cells adopt a survival mode and no longer function normally.
Proteolysis may be activated by inflammatory factors and immobility. The ubiquitin–proteasome and autophagy lysosome pathways are particularly important, contributing to muscle atrophy. These pathways also serve as quality control checks, and so damaged organelles are no longer broken down and recycled.
Ion channel dysfunction, particularly sodium channels, leading to depolarisation and inexcitability of nerve and muscle, potentially through bioenergetic failure.46 49 51 52
Neurophysiological assessment of possible ICU-acquired weakness
Table 3 summarises the neurophysiological features of critical illness polyneuropathy, critical illness myopathy and prolonged neuromuscular blockade. Nerve conduction studies are difficult in ICU, encumbered by the same obstacles facing neurologists; it is tricky to measure a small sensory action potential owing to machinery interference and frequently patient oedema. Critical illness polyneuropathy is an axonal sensory and motor neuropathy.
The neurophysiological assessment of ICU-acquired weakness has a few peculiar features, including:
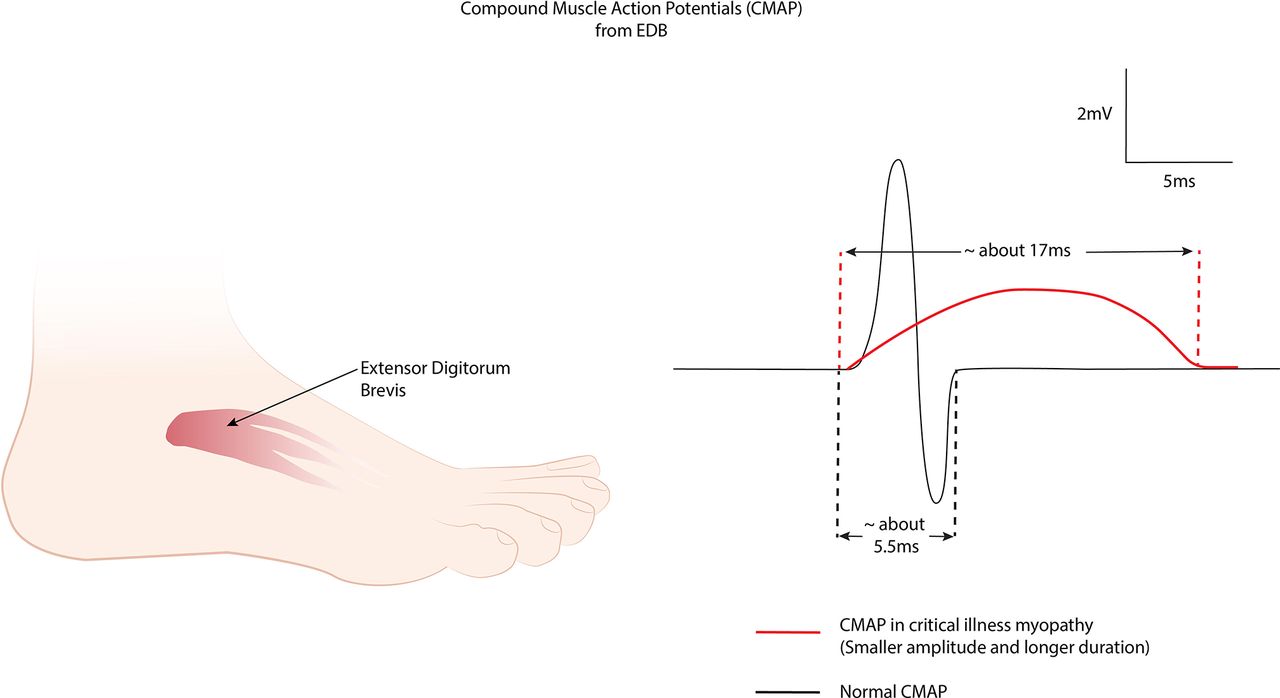
Direct muscle stimulation. The muscle is directly stimulated and the response recorded by electrodes placed within the muscle belly. The muscle response is also recorded conventionally by nerve stimulation, see figure 9.53 A ratio of nerve stimulation to direct muscle stimulation above 0.5 favours critical illness myopathy, whereas a ratio below 0.5 favours critical illness polyneuropathy. This simply reflects excitation through the damaged motor nerve, which is unaffected in critical illness myopathy. Beware and always consider this result in the appropriate clinical context, since normal muscle has the same ratio as critical illness myopathy, see figure 9.
The duration of the compound muscle action potential response in critical illness myopathy is longer than in either normal subjects or in critical illness polyneuropathy, reflecting the slower conduction along myofibres, secondary to ion channel dysfunction, see figure 10.54

Direct muscle stimulation (DMS) and nerve stimulation (NS) of tibialis anterior. A ratio of NS/DMS distinguishes critical illness myopathy from critical illness polyneuropathy.

Prolonged compound muscle action potential (CMAP) in critical illness myopathy, showing a long negative phase without a terminal positive phase.
Muscle biopsy
A muscle biopsy in critical illness polyneuropathy shows denervation.
Critical illness myopathy has three subtypes identifiable on biopsy29:
Loss of thick filaments, perhaps reflecting amino acid diversion towards gluconeogenesis or acute-phase protein synthesis in the liver during critical illness.
Cachectic myopathy.
Necrotising myopathy.
Histopathology in critical illness myopathy therefore ranges from normal despite weakness—reflecting excitability changes in the sarcolemma that leaves no histological footprint—to selective loss of thick myosin filaments, widespread myonecrosis or severe type 2 muscle fibre atrophy.
Personally, therefore, I do not request a muscle biopsy but find neurophysiology helpful.
Serum creatine kinase and CSF
CSF is normal and serum creatine kinase varies significantly, usually being normal or slightly elevated. Even when there is muscle necrosis, the rise in creatine kinase can be transient, and easily missed with a one-off reading. If the creatine kinase is significantly elevated then other conditions need to be entertained, including some in tables 1 and 2.
Consequences of ICU-acquired weakness
These include:
Higher mortality, compared with other hospital patients and ICU patients during the illness and at 1 year.55
Prolonged weaning and reintubation. Patients may require reintubation as late as 2 days following extubation. In people with neuromuscular respiratory weakness, the weakened diaphragm is working near to its maximal transdiaphragmatic pressure and the duration of inspiratory time relative to total breathing cycle duration increases to maintain ventilation. Diaphragmatic fatigue, decreasing strength in response to this contractile activity, is therefore more likely.17 56 57
Increased ICU length of stay is especially likely with critical illness polyneuropathy, where the duration of ICU stay averages roughly 40 days, two times that of critical illness myopathy (20 days).47
Long-term disability. Even those who make a good recovery may have persistent sensory loss, muscle atrophy, pain and focal weakness such as foot drop.49
Preventing and treating ICU-acquired weakness
Unfortunately, there are no specific treatments for ICU-acquired weakness but some measures may help to prevent it. These include:
Controlling plasma glucose with insulin reduces the incidence of ICU-acquired weakness, the duration of mechanical ventilation and of ICU stay, and mortality at 180 days58
Postponing parenteral nutrition to beyond the first week (perhaps counterintuitively) protects against ICU-acquired weakness; it remains uncertain when it is optimal thereafter to introduce parenteral nutrition.
Sparing sedation, including having daily interruptions of sedative infusions.46
ICU philosophy now encourages early mobilisation with passive and active exercises, but we still need more clinical trials and must remain aware of the potential pain that patients feel.2 59
Approximately a third of people with ICU-acquired weakness die in the acute phase, a third become ambulant within 4 months but for many this takes considerably longer. Many are left with sensory loss, atrophy or focal weakness.29 49 60
Other causes of acquired weakness during ICU admission
Stroke, including border-zone stroke, develops in 1%–4% of patients with non-neurological conditions admitted to ICU.61 Stroke in cardiac ICU is normally attributable to embolisation from aortic atheroma.
Border zones are areas of the brain and spinal cord that are furthest from a main arterial supply; they become susceptible to ischaemia with even subtle but sustained periods of hypotension and are often bilateral.
Internal border-zone infarcts usually result from isolated hypoperfusion; these develop at the junctions between the distal branches of the main cerebral arteries (anterior, middle and posterior) and the deep perforating arteries (such as lenticulostriate or anterior choroidal arteries). Cortical border-zone infarcts more often reflect a combination of hypoperfusion and an embolic source, with impaired washout of emboli in these low flow areas.
Examples of border-zone infarctions on ICU include:
‘Man in a barrel’. One border zone in the motor homunculus represents the shoulder, arm and thigh, whereas representation of the foot falls in the anterior cerebral artery, and that of the face within the middle cerebral artery. Bilateral border-zone infarction therefore results in bilateral arm weakness, sparing the legs and face, the so-called ‘man-in-a-barrel’.62
Thoracic paraparesis with dissociated sensory loss. Another border zone is the mid-thoracic spinal cord, relying mainly on the artery magna of Adamkiewicz; this usually arises from the left T10, T11 or T12 intercostal artery, replenishing and sustaining the anterior spinal artery. Bilateral border-zone infarction here leaves the legs flaccidly weak with a truncal sensory level to pain and temperature, usually sparing dorsal columns. Spasticity develops later, sometimes with leg atrophy and fasciculation, reflecting anterior horn cell loss.
A monoparesis may reflect stroke but more commonly results from a pressure palsy: the ulnar nerve at the elbow, the peroneal nerve at the fibular head, or the radial nerve in the spiral groove. Positioning and weight loss both increase the risk. I recently encountered a more unusual and very disabling mononeuropathy that developed in a patient following a prolonged ICU admission for Guillain-Barré syndrome, see figure 11.

High median nerve palsy complicating prolonged intensive care unit admission. Wasting of the thenar eminence with loss of tonic activity means the thumb rests parallel to other digits (‘simian hand’) (left image); it should sit ventrally. The patient cannot pinch (anterior interosseus nerve) but also has weakness of flexor digitorum superficialis index finger when making a fist, almost but not quite a complete ‘benediction sign’ (right image).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS). There is parieto-occipital cortical and subcortical white matter hyperintensity with mild effacement of sulci (MR axial FLAIR) not conforming to a single arterial territory. FLAIR, fluid attenuated inversion recovery.
Central pontine myelinolysis
Central pontine myelinolysis follows rapid changes in serum sodium and osmolarity and may develop in ICU. These patients have a flaccid tetraparesis from corticospinal tract damage in the basis pontis; spasticity emerges later. Importantly, the prognosis is good.63
Wernicke’s encephalopathy frequently accompanies central pontine myelinolysis (30% in pathological series) and up to 43% of ICU patients have vitamin deficiency, another potential neuropathy that develops on ICU.64
When the cause of weakness remains obscure
Despite our best efforts, sometimes it is still difficult to establish why a patient on ICU is weak. Neurologists then need a practical systematic approach, starting with a review of the patient’s medication and of simple blood tests, including serum creatine kinase, calcium, magnesium, phosphate and potassium. MR of the brain and spine should follow, if necessary, and then a neurophysiology assessment if imaging is unremarkable.65
Further reading
Maramattom BV, Wijdicks EF. Acute neuromuscular weakness in the intensive care unit. Crit Med Care 2006;34:2835–41.
Vanhorebeek I, Latronico N, Van den Berghe G. ICU-acquired weakness. Intensive Care Med 2020;46:637–653.
Key points
A neurologist’s clinical assessment of respiratory muscle and spirometry can be life-saving, identifying patients with significant respiratory weakness long before they develop hypoxaemia and hypercapnia that presage impending respiratory arrest.
Good communication with intensive care unit (ICU) colleagues is key, particularly at the thresholds between ICU admission and ICU discharge—jointly assessing patients with ICU colleagues can be very helpful.
The most common causes of new acute-onset respiratory and/or oropharyngeal weakness are Guillain-Barré syndrome and myasthenia gravis, but several other easily overlooked causes include hypokalaemia or hypophosphataemia.
A different range of conditions may present with acute-on-chronic conditions; there are many potential clinical clues but particularly important are motor neurone disease and myotonic dystrophy type 1.
Weakness developing during an ICU admission is common and often reflects ICU-acquired weakness; critical illness myopathy is the most likely explanation, often presenting as difficulty weaning from ventilation, and neurophysiology can help in making the diagnosis.
Supplementary video
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
I thank Siân Morgan, Trainee Clinical Photographer, Medical Illustration, Morriston Hospital and my colleagues Professor Marguerite Hill for her helpful discussion about the patient with myasthenia and Professor Owen Pickrell for figure 12 and Dr Rachel Smith for figure 5.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JW is solely reponsible for this work.
Funding The author has not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed by Robin Howard, London, UK.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Other content recommended for you
- Weakness on the intensive care unit
- Neuromuscular disease and respiratory failure
- Approach to critical illness polyneuropathy and myopathy
- Five-year outcome of respiratory muscle weakness at intensive care unit discharge: secondary analysis of a prospective cohort study
- Respiratory aspects of neurological disease
- First results about recovery of walking function in patients with intensive care unit-acquired muscle weakness from the General Weakness Syndrome Therapy (GymNAST) cohort study
- Mimics and chameleons in Guillain–Barré and Miller Fisher syndromes
- Proximal diabetic neuropathy presenting with respiratory weakness
- Weaning from mechanical ventilation in people with neuromuscular disease: protocol for a systematic review
- Respiratory involvement in inherited primary muscle conditions