Article Text

Abstract
The progression of motor neurone disease (MND) is currently irreversible, and the grave implications of diagnosis naturally fuels concern among neurologists over missing a potential mimic disorder. There is no diagnostic test for MND but in reality there are few plausible mimics in routine clinical practice. In the presence of a progressive pure motor disorder, signs such as florid fasciculations, bilateral tongue wasting, the ‘split hand’, head drop, emotionality, and cognitive or behavioural impairment carry high positive predictive value. MND is clinically heterogeneous, however, with some important chameleon-like presentations and considerable variation in clinical course. Lack of confidence about the scope of such variation, or an approach to diagnosis emphasising investigations over clinical common sense, has the potential to exacerbate diagnostic delay in MND and impede timely planning of the care which is essential to maximising quality of life.
- MOTOR NEURON DISEASE
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
There can be few worse tasks facing the neurologist than giving the diagnosis of motor neurone disease (MND), a condition which is not well understood by the general public.1 Thus, the news of relentlessly progressive limb weakness, likely involvement of speech, swallowing and breathing, dramatic shortening of life expectancy, but no significant disease-modifying therapy, is as surprising as it is devastating for the patient and their family. The Oxford neurologist W Bryan Matthews perfectly articulated the unique challenge of MND for the clinician:
The best test of a physician's suitability for the specialized practice of neurology is not his ability to memorize improbable syndromes but whether he can continue to support a case of motor neurone disease, and keep the patient, his relatives and himself in a reasonably cheerful frame of mind.2
It is easy to understand why the neurologist might wish to put off conveying the diagnosis and to undertake every possible investigation in the pursuit of treatable alternatives. Might we, as neurologists, also occasionally succumb to the fear of just not wanting to get it wrong in a speciality where diagnostic precision is so highly prized? Most MND mimic disorders are equally irreversible, though there are still implications for the accurate counselling of patients and care-planning, meaning it is entirely appropriate to pursue these vigorously where clinically indicated. The diagnosis of MND is only very rarely reversed in favour of a treatable disorder and, in our experience, the reaction of the patient in this situation is one of relief not hostility. Conversely, significant distress can arise from avoidable diagnostic delay and this may permanently erode confidence in onward management.
There is no diagnostic test for MND, and the quest for biomarkers is ongoing.3 Diagnostic delay in MND due to the insidious nature of disease onset and referral from primary care to inappropriate specialists are to a large extent beyond the influence of the neurologist (figure 1). The most robust marker of prognosis is still the interval from symptom onset to definitive diagnosis, the average for which has remained approximately 1 year,4 around a third of the median survival from symptom onset.5 More aggressive and classical MND is referred more quickly to neurologists;6 thus, true mimics (and also some of the chameleons) tend to be slowly progressive disorders and time is often the neurologist's most valuable diagnostic tool.
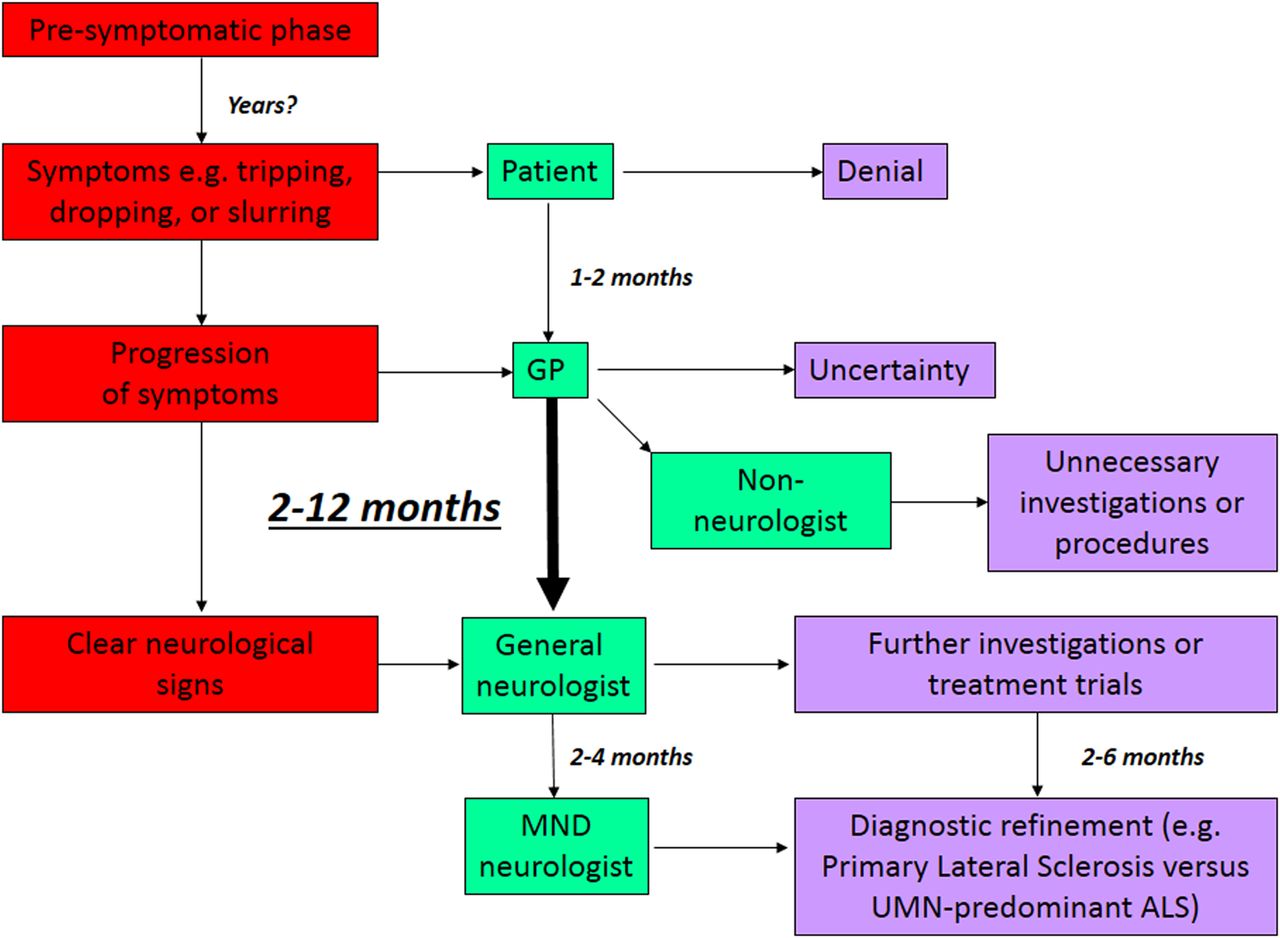
The diagnostic pathway in motor neurone disease (MND). There is an average delay of 1 year between symptom onset and diagnosis. There is a powerful relationship between rate of progression and speed of diagnosis. ALS, amyotrophic lateral sclerosis; GP, general practitioner; UMN, upper motor neurone.
The history
The difficulty increased so that she dragged the right leg, which felt heavy and cold, and it commenced to waste. The right arm began to fail…gradually progressive weakness came on, just like the leg, but especially in the thumb and forefingers, and she experienced a difficulty in holding things. Sensation in no way affected... The wasting progressed daily until she became a living skeleton unable to move, and the breathing was mostly abdominal.7
Sir Frederick Walker Mott's 19th century case report history is inescapably that of MND, even down to the now recognised patterns of spread of symptoms to contiguous body regions.8 ,9 The ‘core’ features of the history are:
-
Steadily progressive initially typically asymmetric weakness (first affecting the lower limb in 35%, upper limb in 30%, and speech and swallowing in 30%)
-
Absence of significant sensory symptoms.
The common perception that MND patients are premorbidly ‘fitter’ or more athletic remains attractive but unproven.10 There is currently no convincing evidence for an MND premorbid personality type.11
The examination
There is great wasting of the muscles of the upper and lower limbs and trunks, especially the small muscles of the hands. Fibrillary contraction may be observed in the limbs and trunk. Elbow tap exaggerated on left side, and also on right, but much less marked. Knee-jerk exaggerated on both sides.7
The physical signature of ‘classical’ MND is captured once again, but importantly only in the context of a prior history of progressive weakness. The core features here are:
-
Upper motor neurone (UMN) and lower motor neurone (LMN) signs in the same territory
-
Florid fasciculations.
There are a handful of key signs on examination that, in the presence of a history of progressive weakness in someone aged over 50 years, should immediately prompt consideration of the diagnosis of MND (table 1). However, the great challenge of MND is that it is evidently a syndrome, and the relative degree of UMN and LMN predominance is highly variable and influences prognosis. At least 10% survive into a second decade, over-represented by those with either ‘pure’ LMN or UMN involvement.12 By definition, classical amyotrophic lateral sclerosis (ALS) displays evidence of both UMN and LMN involvement, accounting for 85% of cases of MND, so that the terms are largely synonymous. Furthermore, many of those with apparent LMN-only disease, sometimes termed progressive muscular atrophy, have subclinical evidence of UMN pathology,13 so the distinction is not particularly meaningful. The very rare (<2%) patients with a pure UMN syndrome, termed primary lateral sclerosis, are characterised by slow progression,14 but may be difficult to distinguish from the so-called UMN-predominant forms of ALS15 (see below). Within this taxonomy, the site of symptom onset also varies. There is also a clear clinicopathological overlap between MND and frontotemporal dementia. Although most cases of MND show only mild or no cognitive impairment, up to 15% may present with frank frontotemporal dementia, which is then associated with more rapidly progressive motor involvement.16
Signs with a high positive predictive value* for motor neurone disease where there is a history of progressive motor-only weakness
Mimics: other causes of progressive motor-only weakness
Population-based studies have indicated that nearly 10% of patients who are diagnosed with ALS ultimately turn out to have another condition (see table 117), but only a few conditions account for the majority of these misdiagnoses. Mimics can be grouped into those presenting with LMN- or UMN-only signs and those with mixed signs. LMN presentations represent the greatest diagnostic challenge. Mimics are listed below in estimated order of frequency in our tertiary referral clinic setting. Key ‘red flags’ can be identified for each of these (table 2). Conditions with sensory involvement as a core feature, for example, syringobulbia, are not considered.
Main mimics of MND based on experience of approximately 1000 patients seen in a tertiary referral clinic, and with the key clue to alternative diagnosis listed
LMN-only signs
Benign fasciculations
The anxious medical student, qualified clinician18 or other individuals (typically under 40 years of age) presenting with fasciculations is a common clinical encounter. While ‘muscle twitching’ was the most sensitive symptom-based keyword for internet search engines to suggest MND as a diagnosis,19 it is part of normal physiology. Exercise, anxiety, caffeine and alcohol may all provoke fasciculations, and approximately 10% of thyrotoxic patients display them floridly.20 It is our experience that those with MND typically do not perceive their fasciculations until pointed out by the physician or a partner and so, in contrast to benign fasciculations, they are rarely the presenting complaint.
In the absence of weakness or abnormalities of thyroid function or electrolytes, individuals aged less than 40 years can be reassured without resorting to electromyography (EMG) to avoid the small but highly damaging possibility of false-positives. Equally, however, most subspecialists will recall a small number of cases, typically men in their 50s or 60s, in whom the latency from presentation with apparently benign fasciculations to weakness (and then clear MND) was several years. Our impression is that a clue may be that the fasciculations of MND are often abrupt and widespread at onset in an individual previously unaffected by fasciculations in youth. The site of the fasciculations, for example, those in the calves versus abdomen, has not been shown to be discriminatory for a benign disorder. There is conflicting evidence as to whether the character of fasciculations differs neurophysiologically in MND.21 ,22 EMG is only 60% sensitive for MND, however,23 and should not be treated as a diagnostic test, but carefully weighed up in the context of the clinical features.
Cramps and stiffness are a frequent co-occurrence with otherwise benign fasciculations; in the absence of weakness, these may be labelled as cramp-fasciculation syndrome, which improves with carbamazepine. True neuromyotonia (Isaacs’ syndrome) is an extremely rare cause of fasciculations,24 and serum antivoltage gated potassium channels are only 50% sensitive. The absence of weakness in both cases is the most useful reassuring feature.
Multifocal motor neuropathy with conduction block
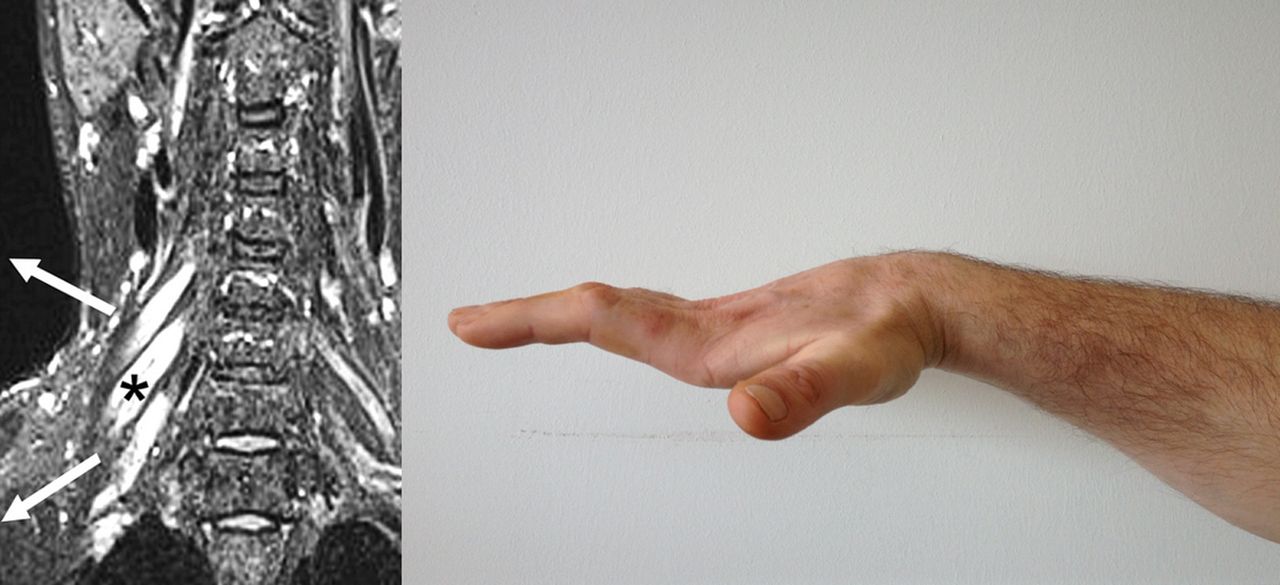
Multifocal motor neuropathy with conduction block has a prevalence of only 0.6/100 000,25 which is 10 times rarer than MND. It is a pure motor neuropathy characterised by slowly progressive, asymmetrical and distal weakness. There is a male predominance (3:1 compared with 3:2 in MND), and a younger age at symptom onset (mean 40 years compared with 65 in MND), with no cases reported aged over 70 years. Wrist or finger drop are common initial symptoms, but a third of cases present in the lower limb. Weakness with very little wasting is the general rule, certainly in early disease, and this should always prompt consideration of an alternative diagnosis to MND. Reflexes are usually lost but can be retained, and even brisk in up to 20% of cases. The absence of bulbar involvement is a notable feature. Respiratory involvement is exceptional and limited to rare case reports of phrenic nerve involvement. Vocal cord paralysis occasionally occurs (personal communication—Dr Chris Allen, followed by personal experience—MRT), but overall multifocal motor neuropathy with conduction block is very slowly progressive and associated with a normal life expectancy. Demonstration of conduction block by an experienced neurophysiologist is diagnostic but not always detectable. Positive anti-GM1 ganglioside antibodies (though only 50% sensitive) and T2-hyperintensity of the brachial plexus (also low sensitivity, figure 2) may be helpful adjunctive tests. Early intervention with intravenous immunoglobulin may limit axonal loss, and so rate of progression.
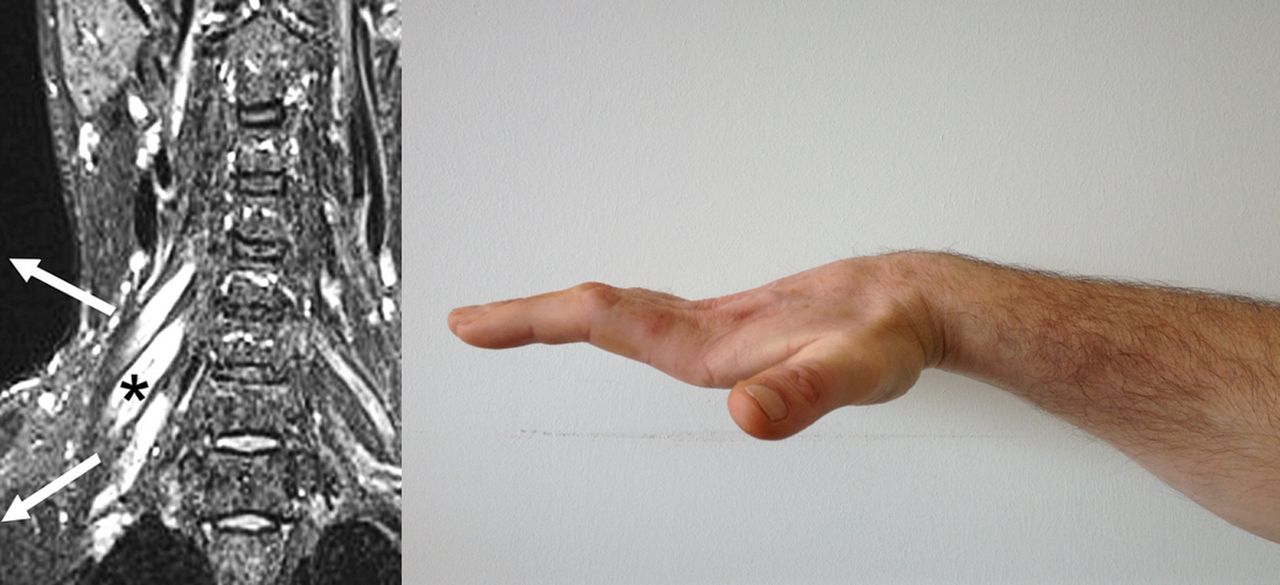
Multifocal motor neuropathy with conduction block. Hyperintense swollen right-sided nerve roots on cervical spine coronal MRI (left panel, modified with permission from71). It typically causes asymmetrical distal weakness, often involving finger extensors initially, but can progress to wasting and fixed flexion deformity, as in this patient, several years after symptom onset (right panel; for other images see72).
Neuralgic amyotrophy
The Parsonage–Turner syndrome26 (neuralgic amyotrophy or brachial neuritis) typically presents with a history of severe unilateral neck, shoulder or arm pain in the absence of trauma, followed over weeks by progressive upper limb weakness and muscle wasting, often involving multiple nerve root territories. Up to 5% of cases may be painless, however. Lower limb variants are much less common. There is often a history of preceding viral illness or vaccination. In 10% of cases of neuralgic amyotrophy there is a family history, with half of these linked to a point mutation or duplication in the SEPT9 gene.27 The key to excluding MND is that the process of weakness and wasting clearly arrests, with variable and slow recovery over months to years. In 10% of cases there is a bilateral pathology, and a particular catch is the involvement of the phrenic nerves, producing diaphragm paralysis with orthopnoea.28
Spinobulbar muscular atrophy
Spinobulbar muscular atrophy, or Kennedy's disease, is a rare X-linked disorder (manifest only in men) associated with a trinucleotide repeat in the first exon of the androgen receptor gene.29 ,30 Patients typically develop symptoms in the 4th or 5th decade or life, involving a very slowly progressive LMN syndrome, with muscle cramps and initially weakness and wasting in the legs or bulbar region (figure 3). Fasciculations are common and, when present over the chin, highly suggestive. Reflexes are absent or reduced. There may also be signs of androgen insensitivity with gynaecomastia and reduced fertility. Though Kennedy's disease can result in significant disability in later years, including respiratory involvement, lifespan is typically normal. The genetic test is diagnostic. There is no effective treatment at present. The slow symmetrical progression, and distinctive clinical features, should make the distinction from MND straightforward.
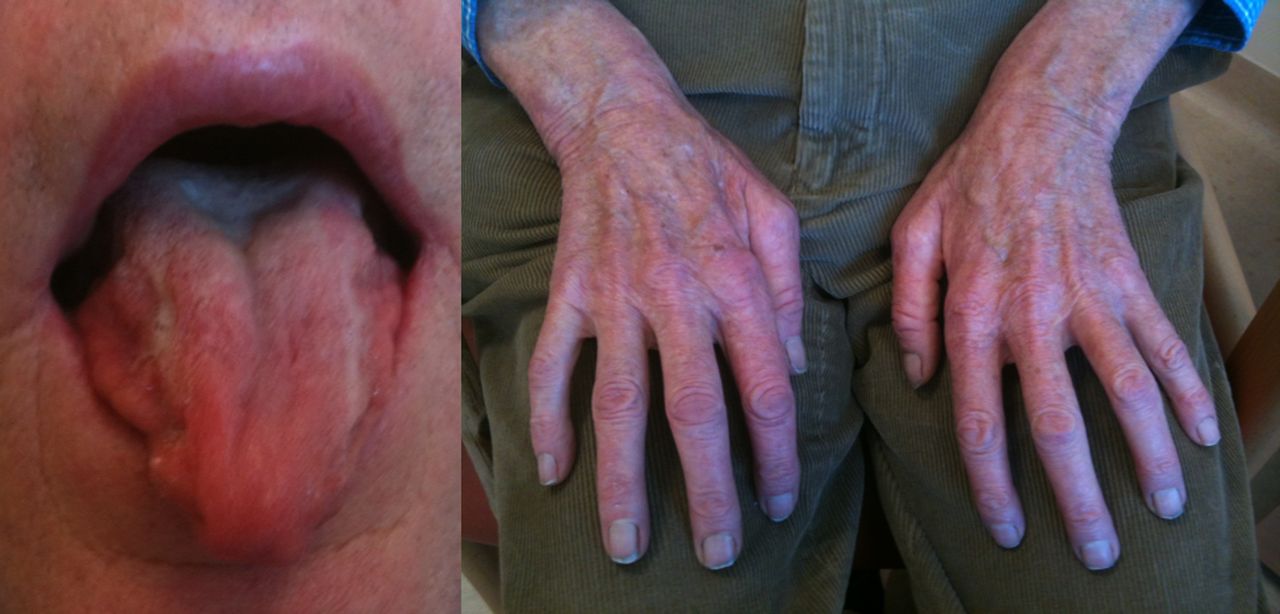
Marked bilateral tongue and hand wasting in Kennedy's disease. The clue to this case not being motor neurone disease was the very slow progression, lack of upper motor neurone signs, gynaecomastia and chin fasciculations.
Motor-predominant chronic inflammatory demyelinating polyradiculoneuropathy
A motor-predominant variant may account for up to a third of all cases of chronic inflammatory demyelinating polyradiculoneuropathy, which has a similar incidence to MND.31 It is distinguished by a relapsing and remitting course, and is predominantly symmetrical with clear demyelination electrophysiologically. There is a notable lack of response to corticosteroids, which favours initial treatment with intravenous immunoglobulin (box 1).32 ,33
Features that may support a trial of intravenous immunoglobulin in a case of progressive motor neuropathy
-
Significant abnormalities on nerve conduction studies, for example, conduction block or demyelination
-
Monomelic slowly progressive lower motor neurone presentations, especially distal
-
Anti-GM1 antibody positivity with no definite upper motor neurone signs
-
Significantly raised cerebrospinal fluid protein
-
Evidence of an underlying malignancy without resectable tumour
Inclusion body myositis
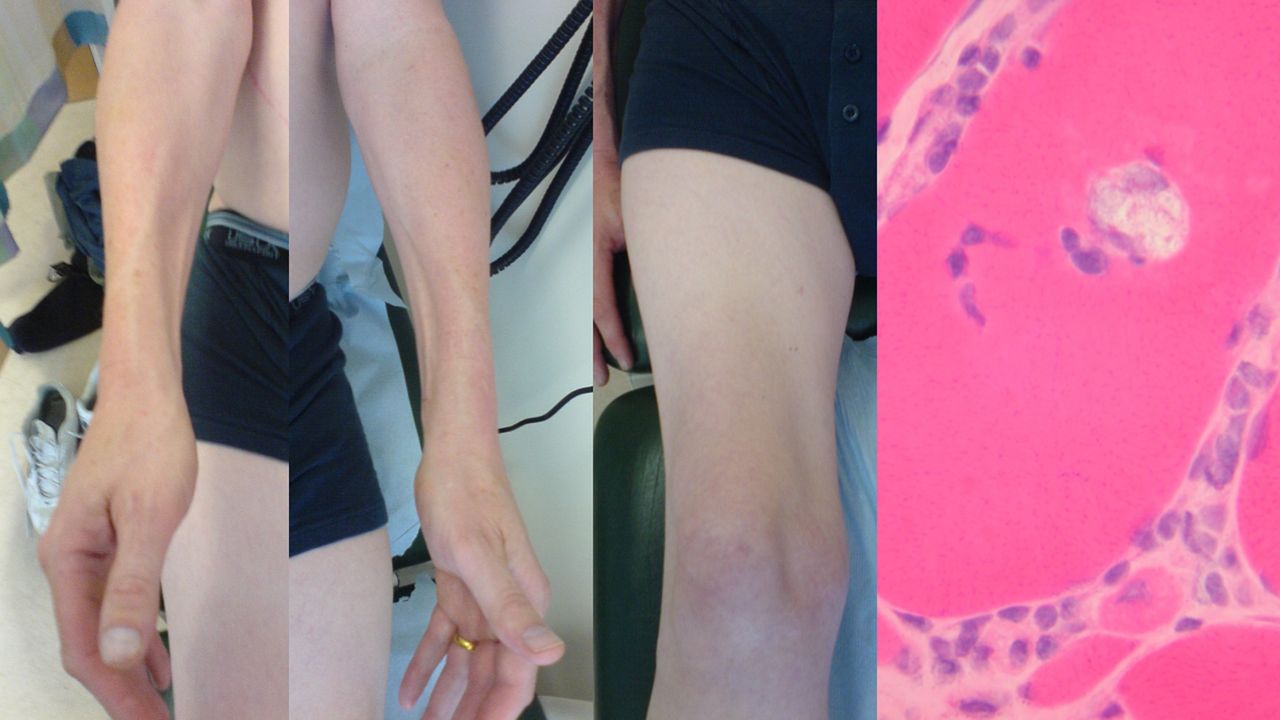
Inclusion body myositis is very much rarer than MND, and is a slowly progressive painless myopathy with a characteristic predilection for wasting of the medial forearm and quadriceps muscles (figure 4). Typically patients are aged over 50 years, with 3:1 male to female ratio and in the overwhelming majority of cases it is an apparently sporadic disorder. At first, there is usually asymmetrical proximal lower limb or distal upper limb weakness.34 Dysphagia occurs in a third of patients and rarely may be the initial symptom. A biochemical clue may be a raised serum creatine kinase beyond what is reasonable for denervation, for example, >1000 IU/l, though it can be normal. EMG is a classic pitfall for the unwary as it can appear neurogenic with fibrillation potentials. The muscle biopsy is diagnostic when it shows characteristic rimmed vacuoles, with inflammatory change (the latter only in sporadic cases). Immunological therapies have not been systematically assessed, and studies report only limited effects.35 The natural history is one of relentless progression over years, with significant disability and probably a reduction in life-expectancy as a result. Interestingly, there are rare hereditary forms of inclusion body myositis, including those associated with both MND and frontotemporal dementia, through mutations in the gene for valosin-containing protein (VCP), and muscle biopsies in inclusion body myositis stain for TDP-43, the characteristic protein aggregating in the nervous system in MND.36 Inclusion body myositis might therefore be best considered a degenerative disorder of muscle.

Typical bilateral medial forearm and quadriceps (right leg only in this case) wasting of inclusion body myositis. There is a characteristic rimmed vacuole within a muscle fibre histopathologically (with acknowledgement to Dr Waney Squier).
Asymmetrical spinal muscular atrophy
Occasionally patients present with asymmetrical, slowly progressive segmental weakness and wasting of the upper limb. Although labelled as ‘asymmetrical spinal muscular atrophy’ this is a purely descriptive term rather than a diagnosis, and there are several underlying causes. Hirayama's disease37 is a characteristic form of upper limb wasting that is seen almost exclusively in young men (16–25 years) from Asia (Japan, the Indian subcontinent and China). One of the most consistent features is the striking unilaterality in most cases, though 20% of patients have evidence of more modest involvement of the contralateral arm. The wasting and weakness characteristically involve muscles innervated by the C7–T1 segments. Thus, the shoulder girdle is usually spared and there is often a striking sparing of brachioradialis (C5,6) in the forearm, giving rise to an appearance which Hirayama called ‘oblique amyotrophy’. Fasciculations appear to be rare. It was shown that progression appears to arrest within a few years and patients followed up for more than 20 years never go on to develop progression to other areas of the motor system.38 In addition to weakness and wasting, patients may describe worsening of their symptoms in cold weather. There is a debate about whether this condition represents a focal form of primary LMN degeneration (ie, a focal form of spinal muscular atrophy) or a local consequence of chronic compression from a dural expansion in the cervical spine (figure 5).

Flexion cervical spine MRI in Hirayama disease showing expansion of the dural venous plexus with presumed chronic ischaemic damage preferentially involving the anterior horn cells supplying the distal arm.
Cervical polyradiculopathy
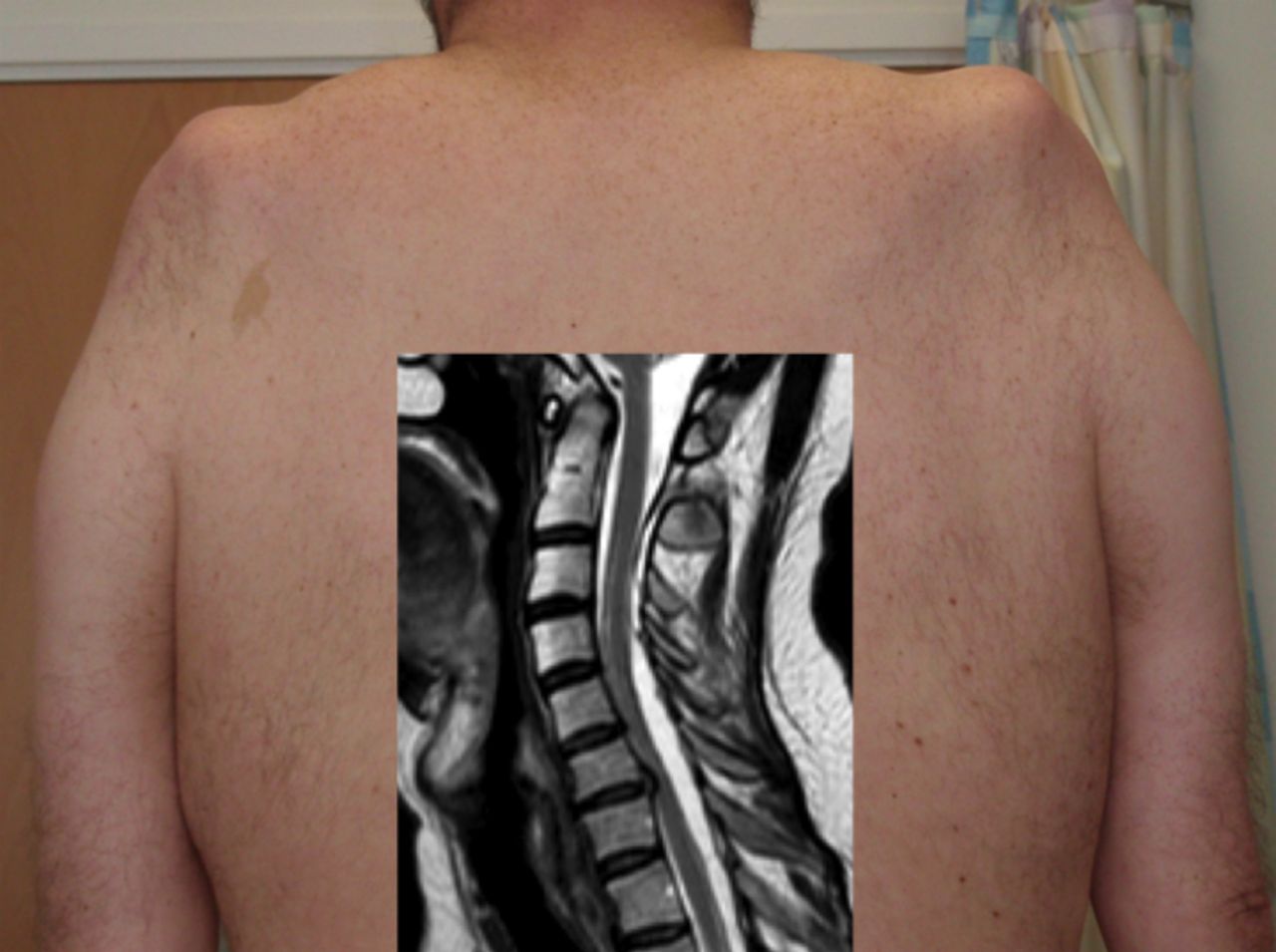
LMN-predominant upper limb presentations of MND frequently involve multiple myotomes, making cervical spine spondylotic polyradiculopathy an unlikely consideration. However, the characteristically slowly progressive, symmetrical LMN ‘flail arm’ variant of MND (also known as brachial diplegia or ‘man-in-a-barrel’ syndrome)39 is one subtype that can be mimicked by cervical spine pathology. Expansion of the dural space, presumed to occur after a previous traumatic cerebrospinal fluid (CSF) leak, results in cord compression and a slowly progressive, symmetrical upper limb proximal weakness with wasting, which may arrest spontaneously, though surgery has been undertaken in some patients (figure 6).40
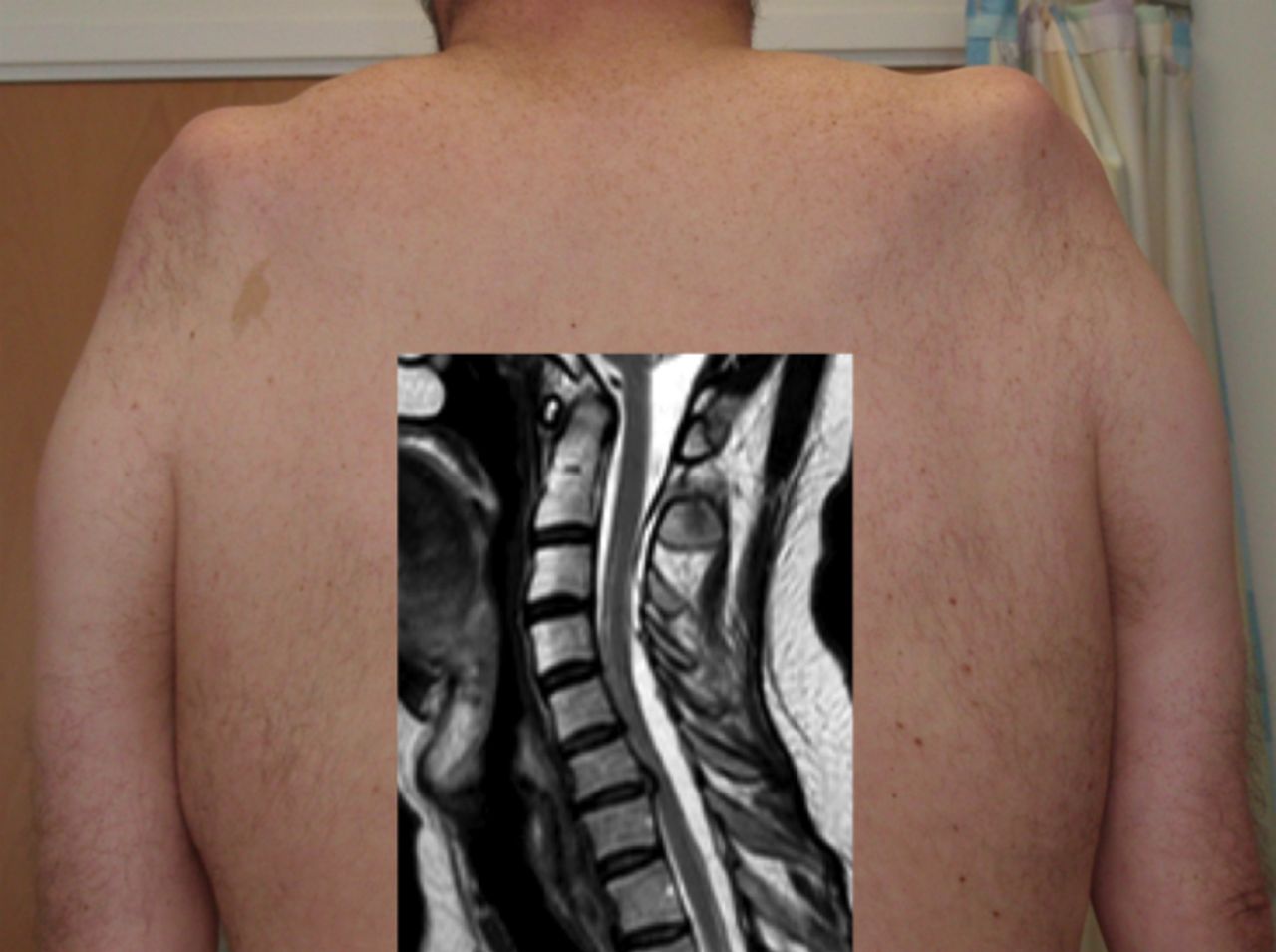
This patient (shown from behind) presented with slowly progressive weakness and wasting of the shoulder girdles, neurophysiological evidence of denervation and slightly raised serum creatine kinase, all compatible with the ‘flail arm’ variant of motor neurone disease. MRI (inset) of the cervical spine showed expansion of the dural space posteriorly with forward compression of the spinal cord and presumed chronic damage to the anterior horns cells. His progression arrested spontaneously.
Radiation-induced radiculopathy
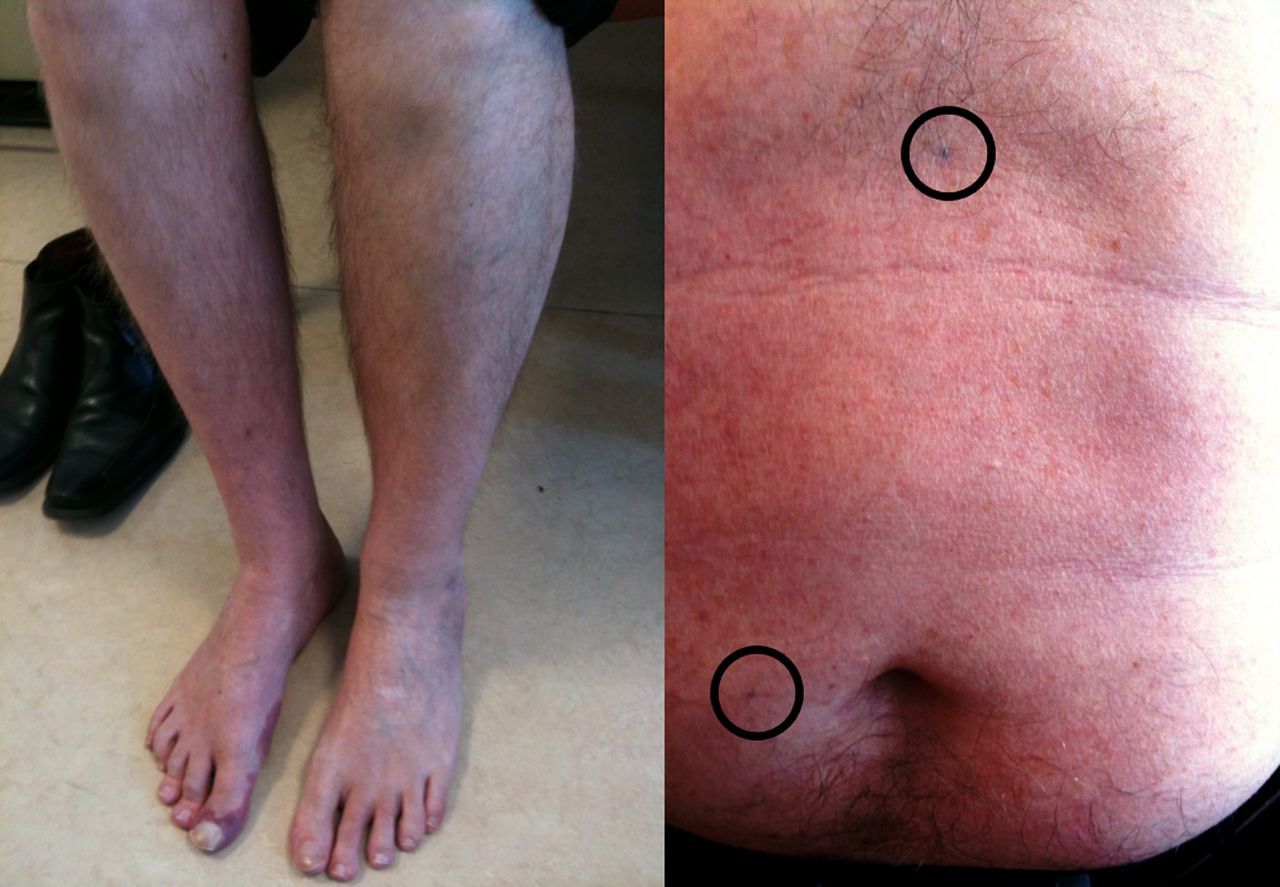
Radiotherapy to the pelvis and para-aortic lymph nodes for testicular and gynaecological tumours may lead to an LMN syndrome which often lacks sensory symptoms. The latency to developing symptoms may be decades (33 years in one case—MRT), and is thought to be mediated by a vasculopathy; thus, the history of exposure must be specifically sought (figure 7).41 The condition is usually very slowly progressive but may lead to significant disability and eventual sphincter involvement. The equivalent brachial plexus syndrome, typically following breast cancer, usually occurs with a more recent history of exposure and is more easily differentiated from MND by the presence of pain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Radiation-induced lumbar radiculopathy. This patient presented with a slowly progressive right leg weakness and wasting more than 30 years after radiotherapy to the right para-aortic nodes for testicular cancer (radiotherapy tattoos shown circled over abdomen).
Infections
With the hope of near global eradication of poliomyelitis, there are few infectious agents that target the LMN. Although poliomyelitis was once common, there is no evidence of anything more than a coincidental relationship when MND develops in someone previously affected by poliomyelitis. HIV has been linked to a very rare distal LMN syndrome,42 but prominent sensory features are typically a clue in HIV-related neuropathies.43
Grand round rarities
Facial-onset sensory motor neuropathy is a very rare neurodegenerative disorder presenting in a slightly younger age group than MND, typically with trigeminal sensory loss, but also associated with wasting, weakness and fasciculation of the tongue and later respiratory muscle weakness.44 It is arguably more a mimic of syringobulbia. Progression is slow and immunomodulatory therapy has no effect.
UMN-only signs
Hereditary spastic paraparesis
Hereditary spastic paraparesis is a term encompassing a genetically diverse group of disorders characterised by very slowly progressive paraplegia, which can sometimes involve dorsal column and sphincter dysfunction, and also the upper limbs. Additional features in complex genetic subtypes may include ataxia or dementia, but they do not include the often severe corticobulbar involvement seen in primary lateral sclerosis. The presence of a family history (typically autosomal dominant) is a major clue to hereditary spastic paraparesis over primary lateral sclerosis, and 40% of such cases will have mutations in the SPAST gene, though establishing a firm genetic diagnosis in the remainder is not currently straightforward.45
Primary progressive multiple sclerosis
Approximately 15% of patients with multiple sclerosis have a slowly progressive clinical course, without remission, and are termed primary progressive.46 Men are over-represented and the age at symptom onset is older (fifth decade). Primary progressive multiple sclerosis is only a plausible alternative to primary lateral sclerosis where there is no marked bulbar dysfunction, with oligoclonal bands detectable in CSF (though this occasionally occurs in cases of MND47), or evidence of demyelination on MRI.
Metabolic myelopathies
Vitamin B12 and copper48 deficiencies are well-recognised causes of slowly progressive myelopathy, but typically have associated sensory impairment. The entity of functional B12 deficiency (with a normal serum level but transcobalamin dysfunction) remains controversial and is probably very rare.49 X-linked adrenoleukodystrophy is a peroxisomal disorder involving mutations in ABCD1 responsible for very long-chain fatty acid transport (and their accumulation in serum is diagnostic).50 Both men and women carriers can develop an adult-onset slowly progressive (10–20 years) myelopathy, typically in the 3rd or 4th decade, confined to the lower limbs, with sensory ataxia and bladder symptoms. There are frequently no abnormal MRI findings, although occasionally secondary Wallerian degeneration results in hyperintense cerebral corticospinal tracts. There is no effective treatment at present.
Corticobasal degeneration
Corticobasal degeneration is an extremely rare extra-pyramidal rather than UMN neurodegenerative disorder, but is occasionally mislabelled as primary lateral sclerosis. The major clue to corticobasal degeneration is unilateral rigidity and bradykinesia (50%).51 The disease course is much shorter than primary lateral sclerosis (mean 7 vs 10–20 years), and the overt cognitive involvement of corticobasal degeneration is not typically seen in primary lateral sclerosis.
Mixed signs
Cervical myeloradiculopathy
The unique anatomy of the cervical spine in which there is close proximity of both UMN and LMN makes degenerative myeloradiculopathy a potential diagnostic pitfall in cases of suspected MND. Moreover, pure motor syndromes and absent sphincter involvement are not uncommon in cervical spondylotic disease. However, incidental spondylosis of the spine is highly prevalent among those with MND, given the mean age of onset of 65 years, and preventing unnecessary orthopaedic surgery is an ever-present challenge.52 Symptoms such as emotionality and abnormal signs above the neck are helpful in avoiding overinterpretation of neck pathology in relation to limb weakness.
Chameleons: atypical presentations of MND
Occasionally, MND presents outside the rough rule-of-thumb of one third upper limb, one third lower limb and one third bulbar in onset, or symptoms remain confined to only one body region for months or even years.
Monomelic involvement
Cases of what subsequently turns out to be MND with symptoms confined for long periods to a single limb are the most challenging diagnostically, especially where there are only LMN signs. This is where a more reliable diagnostic biomarker might have greatest impact by allowing earlier intervention with future therapies. Flail leg as well as arm variants of MND are well recognised and also typically more slowly progressive.53
Isolated bulbar involvement
Bulbar-onset MND patients are commonly referred to the wrong specialist (ENT or TIA clinic) or inappropriately investigated.54 Historically termed progressive bulbar palsy, there is a subset of bulbar-onset MND patients, typically elderly women, in whom there may be rapidly progressive anarthria, often with prominent emotionality, but with limb function preserved for many months, occasionally years.55 EMG, including sampling of the tongue, may be normal and should not delay the diagnosis and timely gastrostomy insertion. Primary lateral sclerosis can occasionally present with bulbar dysfunction, but is characteristically very slowly progressive with UMN signs ultimately generalising.
It is not uncommon for MND patients with bulbar symptoms to report a degree of diurnal variation in their symptoms, and a very few patients may even test falsely positive for serum antiacetylcholine receptor antibodies or have equivocal neuromuscular junction abnormalities on neurophysiological testing. Eye signs are useful in suggesting the diagnosis of myasthenia gravis (or the extremely rare oculopharyngeal muscular dystrophy, most of whom present with ptosis before significant tongue weakness).
Head drop
Progressive weakness of neck extension is an important physical sign, with a very limited differential diagnosis that includes MND high on the list. It should not be attributed to cervical spondylosis.
Respiratory-onset or acute presentation
About 3% of MND patients develop diaphragm weakness as the initial problem, and unfortunately the prognosis is then typically poor.56 Acute admission of advanced cases of undiagnosed MND in respiratory failure with superimposed pneumonia still occurs. Irreversible and unwanted endotracheal intubation may result, and it is also essential to avoid use of depolarising muscle relaxants in those with severe denervation as these may precipitate dangerous hyperkalaemia.57
Frontotemporal dementia at presentation
At least 10% of MND patients develop clinically obvious frontotemporal dementia in population-based studies.16 In such cases, this is typically a very early symptom and the course of the motor involvement is then more rapid. Severely affected patients may present with profound changes in personality (apathy or occasionally disinhibition) or altered food preference (limited repertoire or particular focus on sweet items, often with food-cramming). Characteristically they have limited insight. Frontotemporal dementia cases linked to expansions in the hexanucleotide repeat in C9orf72 (some of whom develop MND too) may have an increased frequency of psychosis.58
Young-onset MND
The range of age at symptom onset in MND is large (17–97 years in our clinic). Younger age of onset (<45 years) accounts for approximately 10% of patients and is typically associated with a slower rate of progression. Juvenile-onset (<25 years) MND-like syndromes are very rare and typically there is a family history.59 However, an aggressive rare form of LMN-predominant MND associated with mutations in the FUS gene with characteristic basophilic inclusions on postmortem histology is particularly associated with symptom onset in the late teens.60 It can present as a sporadic or familial disorder.
Sensory features
While prominent sensory features as part of the presenting complaint should always prompt re-evaluation if MND is being otherwise considered, some sensory nerve involvement in MND is well recognised,61 and pain is not unusual.62
Parkinsonian features
The ALS-Parkinsonian-Dementia complex of Guam (and the similar disorders in the Kii Peninsula of Japan and Papua New Guinea) represent extreme examples of the phenotypic overlap of Parkinson's disease and ALS. On Guam at least, this seems to be an environmentally-determined disorder in a population with a genetic bottleneck, and is now declining in incidence. However, it has been long recognised that more typical cases of MND often have overlap with Parkinsonism.63 Such features often occur late rather than at presentation, including in patients with primary lateral sclerosis where the combination with bulbar dysfunction and common postural instability may lead to the erroneous diagnosis of progressive supranuclear palsy. A variety of eye movement abnormalities, including supranuclear gaze palsy, have also been described in MND,64 but these are rarely prominent.
Hemiparesis (Mills’ syndrome)
A rare, very slowly progressive spastic hemiparesis with generalised hyperreflexia and no sensory involvement was first described by the US neurologist Charles Karsner Mills (1845–1931). Standard MRI is normal. A positron-emission tomography study demonstrated microglial activation in the contralateral motor cortex,65 and the condition is considered to be a variant of primary lateral sclerosis.
Myths and legends
Textbook lists of MND mimics may be lengthy, but many entries do not seem to reflect routine clinical practice and so have the potential to fuel diagnostic delay, based as they are on a few or even single case reports (nostra culpa 66). All but the most advanced cases of MND will require some investigation, but the concept of ‘mandatory’ investigations might be more rationally stated as just those tests justified by the clinical history and signs, EMG included (table 3).67
Key scenarios when specific investigations have most value in suspected motor neurone disease. None of these investigations is mandatory and are always guided by the clinical features
Paraneoplastic MND
It may seem remiss not to have included paraneoplastic neuropathies as mimics, and certainly these very rare disorders may match MND in their aggressive progression and associated weight loss. However, there are typically prominent sensory as well as motor features, and individuals lack the systemic well-being that is so striking among the vast majority of those with MND, in our experience. Nonetheless, there have been MND-like syndromes linked to lymphoma68 and breast cancer (where it is then typically UMN-predominant69). Lumbar puncture in suspected paraneoplastic disorders should include testing for oligoclonal bands, though these do occasionally occur in classical MND.47
Lyme disease
Although frequently termed the universal neurology mimic, neither acute Lyme disease nor the more controversial entity of chronic neuroborreliosis is convincing as a mimic of MND.70
Lead poisoning and porphyria
The development and pattern of progression of motor neuropathies caused by chronic lead exposure or repeated attacks of acute porphyria are not reminiscent of MND.
Key points
-
Motor neurone disease (MND) is a clinical diagnosis and there are no mandatory investigations.
-
An earlier diagnosis allows patients to maximise the quality of their remaining life and improves care-planning.
-
A progressive motor syndrome with mixed upper and lower motor neurone signs in more than one body region has few, if any, differential diagnoses.
-
Lower motor neurone-predominant monomelic clinical sub-types of MND present the greatest diagnostic challenge but are frequently more slowly progressive.
-
Bulbar-onset MND is not uniformly associated with a rapid progression, and symptoms may remain isolated to this territory with apparently normal EMG studies.
-
MND overlaps pathologically with frontotemporal dementia and around 10% of MND patients develop pronounced cognitive impairment (typically as an early feature).
-
Paraneoplastic MND-like syndromes are extremely rare.
References
Footnotes
-
Correction notice This article has been corrected since it was published Online First. The second of the 'Key points' in the box at the end of the article was duplicated. This has since been removed.
-
Contributions MRT conceived and drafted the paper and figures, and is the guarantor of the content. KT edited the paper.
-
Funding MRT receives funding from the Medical Research Council & Motor Neurone Disease Association UK Lady Edith Wolfson Fellowship (G0701923). The Oxford MND Care Centre is funded through a grant from the Motor Neurone Disease Association UK.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed. This paper was reviewed by Chris McDermott, Sheffield, UK.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Other content recommended for you
- Cortical thickness in ALS: towards a marker for upper motor neuron involvement
- Sensitivity of brain MRI and neurological examination for detection of upper motor neurone degeneration in amyotrophic lateral sclerosis
- Motor neurone disease
- Assessment of suspected motor neuron disease
- Motor neuron disease
- Differentiating lower motor neuron syndromes
- A rapidly progressive neuropathy
- Cognitive and behavioural changes in PLS and PMA:challenging the concept of restricted phenotypes
- Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis
- Diagnosing ALS: the Gold Coast criteria and the role of EMG