Article Text

Abstract
The diagnosis of sporadic Creutzfeldt-Jakob disease (CJD) can be difficult, but the real-time quaking-induced conversion (RT-QuIC) assays have made a considerable impact on its clinical diagnosis. This technique exploits the ability of the misfolded pathological form of prion protein (PrPSc) found in cerebrospinal fluid (CSF) to induce conversion of normal PrP to the misfolded form, which subsequently aggregates. The formation of these aggregates of misfolded PrP is monitored in real time using fluorescent dyes. The current sensitivity of CSF RT-QuIC undertaken at the UK National CJD Research & Surveillance Unit is 92% and the specificity is 100%. The interpretation of the RT-QuIC traces is affected by the presence of raised CSF red and white cells counts and elevated total protein concentrations. We recommend that CSF samples for RT-QuIC analysis are clear and colourless with a white cell count of <10 x10^6/L and have a total protein concentration of <1 g/L.
- CSF RT-QuIC
- cerebrospinal fluid
- prion disease
- creutzfeldt-jakob disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Introduction
Creutzfeldt-Jakob disease (CJD) is a fatal neurodegenerative disease belonging to a family of diseases known as transmissible spongiform encephalopathies, which affect both humans and farmed and wild animals. These diseases are associated with the deposition within the brain of an abnormally folded and protease-resistant form (PrPSc) of cellular protein known as prion protein (PrPC). PrPSc is able to bind to PrPC to induce misfolding and the creation of additional PrPSc, which can then induce further PrPC misfolding and subsequent aggregation into fibrils and plaques. Thus small amounts of PrPSc are able to propagate throughout the central nervous system.
CJD is the most common form of transmissible spongiform encephalopathy in humans; 85% of cases are sporadic in nature, with most of the remaining cases being genetic in origin (15%), secondary to mutations in the prion protein gene (PRNP). A very small number of cases are iatrogenic in nature due to the administration of human cadaveric growth hormone in the 1970–1980s or the use of affected dura mater grafts. Variant CJD, acquired through ingesting foodstuffs contaminated by bovine spongiform encephalopathy or related to transfusion of contaminated blood or blood products, is increasingly rare.1
Sporadic CJD is associated with a rapidly progressing dementia and neurological features such as ataxia and myoclonus; death usually occurs within 6 months. The diagnosis can be difficult in the early stages and is made on the presence of a series of clinical features and distinctive abnormalities in supportive investigations, such as triphasic waves on electroencephalogram, high signal in the basal ganglia on MR scan of the brain, and the detection of cerebrospinal fluid (CSF) 14-3-3 and other markers of acute neuronal damage such as tau protein in CSF.2 3 All these investigations are hampered by the fact that they are surrogate markers and as such detect changes that can occur in many other conditions. Therefore, none of these investigations are specific for CJD and none can detect all sporadic CJD (sCJD) subtypes.4 5
Real-time quaking-induced conversion
The characteristic neuropathological hallmark of transmissible spongiform encephalopathies is the presence of PrPSc. There have been many different approaches to try to detect PrPSc; however, these have been hampered by the very small amounts present in body fluids such as CSF and blood, but also by the lack of antibodies that can distinguish PrPSc from PrPC.
Real-time quaking-induced conversion (RT-QuIC) was described in 2010, a new technique to detect small amounts of PrPSc.6 7 This technique exploits the ability of PrPSc to induce PrPC to misfold in a cyclical fashion to form aggregates of PrPSc fibrils. The formation of these aggregates is monitored in real time by their ability to bind a fluorescent dye, namely thioflavin T (figure 1). In brief, CSF from a patient suspected of having CJD is added to a mixture of recombinant PrP (rPrP) and thioflavin T. Any PrPSc present in the CSF binds to the rPrP and induces it to change shape and form fibrils. This process can take up to 30 hours and is known as the lag phase, after which time the fibrils start to aggregate and bind thioflavin T, which starts to fluoresce (figure 2). This fluorescence is monitored in real time. The aggregation kinetics are monitored and show a characteristic sigmoidal curve. The first studies used either full-length hamster rPrP (23-231) or full-length human rPrP and reported sensitivities between 69% and 89% for neuropathologically confirmed sCJD, and specificities between 99% and 100% (table 1).6 8–13
Reported sensitivities and specificities of RT-QuIC in sCJD, using different rPrP substrates.1
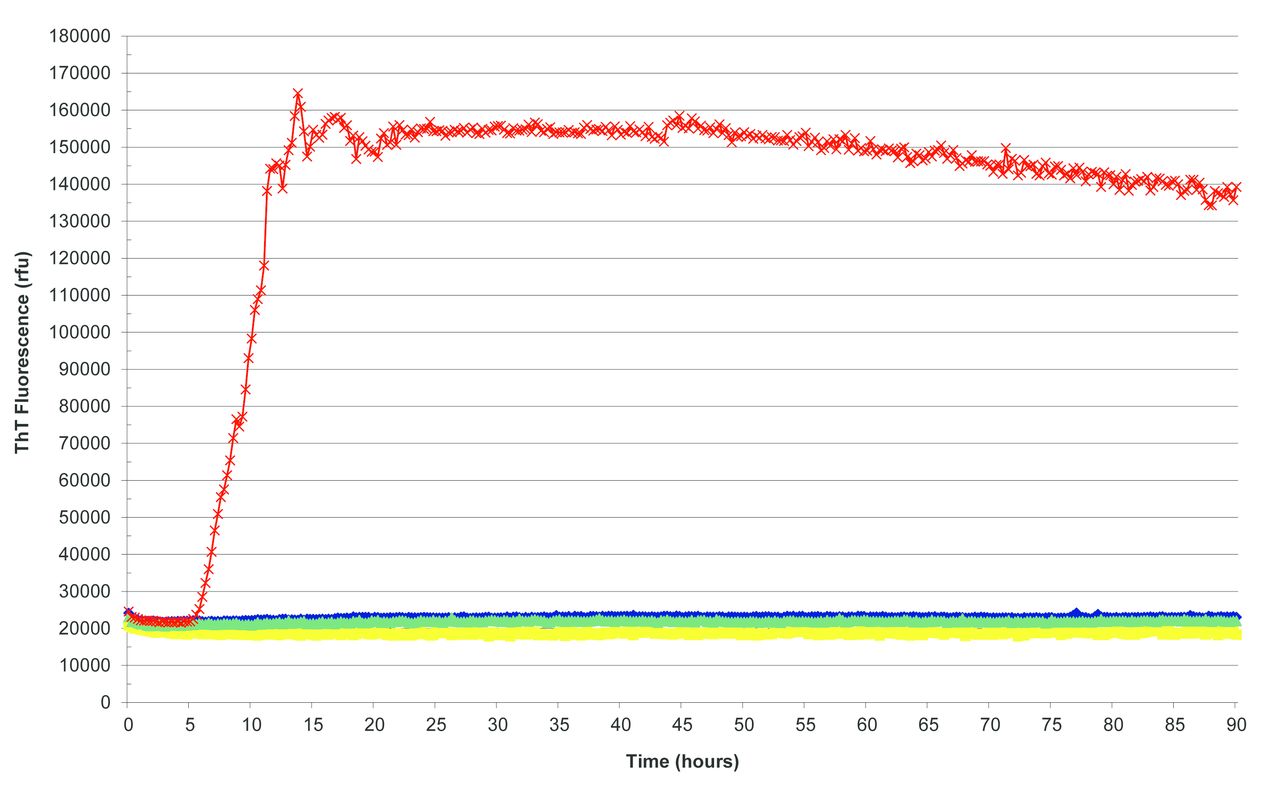
Real-time quaking-induced conversion reactions seeded with sporadic Creutzfeldt-Jakob disease brain homogenate as positive control (red), Alzheimer’s disease brain homogenate (yellow), sudden death brain homogenate from subject with no neuropathological abnormality (green) and unseeded reaction (blue) as negative controls. ThT, thioflavin T.

{kind=link}
{kind=link}
Real-time quaking-induced conversion reactions seeded with cerebrospinal fluid from a patient with sporadic Creutzfeldt-Jakob disease (sCJD) (red) and a patient without sCJD or related prion disorder (blue). ThT, thioflavin T.
In addition to using different forms of rPrP, these studies also used different instrumentation and slight variations in methods. To ensure that there was concordance among these different techniques, we undertook a quality assurance assessment. Two sets of identical CSF samples, containing CSF samples from patients with either sCJD or another neurodegenerative condition, were sent to 10 laboratories undertaking RT-QuIC analysis and analysed blind to the final diagnosis. There was very good concordance with the first set of samples and complete concordance with the second.14 Therefore, there is a good agreement between laboratories undertaking RT-QuIC analysis worldwide.
Despite the name, RT-QuIC is actually a long process taking 90 hours to reach completion. There is now an adaptation of RT-QuIC known as either improved RT-QuIC (IQ) or second-generation RT-QuIC using truncated hamster rPrP (90-231) as substrate. This has the same order of sensitivity (70%–96%) as the traditional RT-QuIC but a much shorter analytical time of approximately 30 hours.11 13 15–17 The specificity of this substrate is 98%–100% (table 1). There is currently a worldwide comparative study of both the first-generation and second-generation RT-QuIC techniques under way.
A direct comparison of RT-QuIC with CSF 14-3-3 and tau protein has shown that RT-QuIC has greater sensitivity and specificity than either CSF 14-3-3 or tau protein.8 12 16 18
Clinical and analytical factors affecting RT-QuIC affecting performance
Sporadic CJD subtypes
Sporadic CJD is a heterogeneous disease with six clinicopathological subtypes based on the PRNP codon 129 genotype: homozygous for methionine (MM) or valine (VV) and heterozygous for methionine and valine (MV), and the type of PrPSc accumulating within the brain (type 1 or 2). Most sCJD cases are MM1, MV1, MV2 or VV2, with MM2 and VV1 cases rarer.19 The sensitivity of CSF RT-QuIC is not influenced by PRNP codon 129 alone (table 1), but CSF RT-QuIC does appear to have a lower sensitivity in patients with sCJD-MM2 and sCJD-VV1 (table 2). We do not know the significance of this at present as there are only small numbers of cases involved, but its significance may become clearer with more cases analysed.
Sensitivity of CSF RT-QuIC in different sCJD subtypes
Age at onset of disease, disease duration and timing of lumbar puncture
The detection of CSF 14-3-3 is influenced by the age at disease onset, disease duration and timing of lumbar puncture, with the highest sensitivity in patients with shortest disease duration, older than 40 years old and in the later stages of disease.20 21 It is less clear whether these factors influence CSF RT-QuIC positivity. McGuire et al8 reported no influence of age at onset of disease, stage of disease or disease duration; however, Cramm et al22 noted that patients with a shorter disease duration had higher seeding activity as measured by a shorter lag phase and higher final fluorescence values. Clearly this needs further investigation, and studies are under way to examine the relationship between CSF RT-QuIC and disease duration or severity at time of CSF collection.
Analytical factors
CSF RT-QuIC is not affected by storage of CSF samples at room temperature or 4°C for up to 8 days nor is it affected by repeated freeze–thaw cycles.23 However, blood contamination in CSF samples does affect RT-QuIC. Red cells in CSF samples inhibit the RT-QuIC response, with fewer replicates giving a positive response, longer reaction times and reduced final responses.16 23 A cut-off of <1250 x 10^6/L has been recommended.23 The haem moiety in haemoglobin is probably responsible for quenching the fluorescent signal, and thereby reducing the RT-QuIC response.
We have also found that high CSF total protein concentrations of >1.0 g/L and raised white cell counts (WCC) may result in an RT-QuIC response that shows a high and fluctuating baseline that could be misinterpreted as a positive RT-QuIC response. As a result, we must interpret CSF RT-QuIC with caution in those samples with elevated total protein or WCC. Although it is unusual for patients with sCJD to have raised total protein concentrations and/or WCC, a small number of patients with sCJD have had CSF total protein concentrations of >1.0 g/L or WCC of >10 x 10^6/L.24
Many laboratories are reluctant to analyse CSF samples from patients with suspected CJD due to the perceived transmission risk. The current guidelines from the UK Department of Health and Advisory Committee on Dangerous Pathogens’ Transmissible Spongiform Encephalopathy subgroup advise that CSF is a low-risk specimen and that no additional precautionary measures are required for biochemical and cytological analysis (part 3 and annex K).25
False-positive CSF RT-QuIC results
In contrast to other CSF biomarkers such as 14-3-3 and tau protein, RT-QuIC has a high degree of specificity in the investigation of sCJD. There are currently eight cases reported in the literature with a ‘false’ positive CSF RT-QuIC results (table 3).8 12 16 22 26 Only three of these cases underwent postmortem examination and one had dementia with Lewy bodies confirmed; however, subsequent western blotting of the brain tissue found small amounts of abnormal PrPSc. This raises the possibility that this patient also had subclinical sCJD. The diagnoses in the remaining two cases were frontotemporal lobar degeneration-TAR DNA-binding protein 43 kDa with motor neurone disease and steroid-responsive encephalopathy presenting with convulsion. In both of these cases, the CSF RT-QuIC traces were reported as slowly and slightly amplifying. A diagnosis of sCJD could not be ruled out in the remaining six cases.
Clinical information and neuropathological and diagnostic investigations in eight patients with ’false’ positive CSF RT-QuIC results
CSF RT-QuIC in other prion diseases
There is less information available on the utility of CSF RT-QuIC in genetic forms of prion disease. However there are increasing data to suggest that CSF RT-QuIC has a high degree of sensitivity in the commoner mutations in the PRNP gene such as E200K (82%–100%) or the insert mutations (100%).12 27 There are fewer data for the rarer mutations. CSF RT-QuIC is not positive in variant CJD, although an alternative form of protein aggregation assay known as protein multiplication cyclic amplification has reportedly been able to detect PrPSc in CSF in variant CJD but not in sCJD; this suggests that a combination of these two techniques may potentially discriminate between these two forms of CJD in life.28 29 In our experience, approximately 67% of patients with iatrogenic CJD secondary to cadaveric growth hormone administration are positive for CSF RT-QuIC.
RT-QuIC using other biological fluids and tissues
As yet, there is no reliable blood RT-QuIC assay for prion diseases for reasons that are outlined above. RT-QuIC analysis of olfactory neuroepithelium nasal brushings has been shown to have a comparable specificity and improved sensitivity when compared with CSF analysis.9 13 However, it is not routine to collect such samples and the technique may require specialist expertise, which limits its usefulness in a clinical setting. A recent report showed that skin samples from patients with sCJD and variant CJD are positive by RT-QuIC analysis, but this has not yet been replicated.30 The skin sample preparation is complex involving homogenisation, phosphotungstate precipitation and ultracentrifugation, and this reduces its practicality as a routine diagnostic test.
Update from the NCJDRSU
Using the first-generation technique8 the National CJD Research & Surveillance Unit (NCJDRSU) has analysed over 1600 CSF samples, including 616 sCJD cases, and our current sensitivity for sCJD is 92% and our specificity is 100%. Of those 616 sCJD cases investigated, 52 were negative for CSF RT-QuIC, and of these 32 were positive for CSF 14-3-3 and an additional 15 cases had other supportive investigations, such as cortical ribboning and/or high signal in the basal ganglia on MRI or triphasic waves seen on EEG. Only five cases were negative for all clinical diagnostic tests and these cases were diagnosed at postmortem.
We are currently undertaking parallel study comparing our current first-generation CSF RT-QuIC assay with the second-generation CSF RT-QuIC assay with the view to moving to the second-generation system provided the same degree of sensitivity and specificity can be guaranteed, thereby reducing the turnaround time by 50%.
Key Points
Cerebrospinal fluid real-time quaking-induced conversion (CSF RT-QuIC) is a highly sensitive and specific test for sporadic Creutzfeldt-Jakob disease (sCJD).
It is not affected by age at onset of disease or PRNP codon 129; however, it may be less sensitive in the rarer forms of sCJD such as MM2 (methionine) with cortical changes and VV1 (valine).
Of those patients who are negative for CSF RT-QuIC, 90% have an alternative positive diagnostic investigation, such as CSF 14-3-3, the presence of cortical ribboning and/or basal ganglia changes on MRI or triphasic waves on electroencephalogram.
The interpretation of CSF RT-QuIC is hampered by the presence of elevated red cell counts (>1250 x 10^6/L), white cell counts (>10 x 10^6/L) and raised total protein concentrations (>1 g/L).
CSF RT-QuIC may be less sensitive in the rarer sCJD subtypes such as sCJD-MM2 associated with cortical changes and sCJD-VV1.
Acknowledgments
I thank Dr Lyne McGuire and Dr Neil McKenzie for their hard work in establishing and undertaking CSF RT-QuIC analysis within the NCJDRSU. In addition, I would like to thank Dr Neil McKenzie for producing the figures within this manuscript. The surveillance of CJD is dependent on the cooperation of neurologists and neuropathologists throughout the UK. The author is particularly grateful to the relatives of the patients for their cooperation. The views expressed in this publication are those of the author and not necessarily those of the Department of Health.
References
Footnotes
Contributors I am the sole contributor to this manuscript.
Funding This study was funded by the Policy Research Program of the Department of Health and the Government of Scotland (Grant Number: PR-ST-0614-00008).
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned. Externally peer reviewed by Simon Mead, London, UK.
Other content recommended for you
- CJD mimics and chameleons
- Clinical features of genetic Creutzfeldt-Jakob disease with V180I mutation in the prion protein gene
- Characteristics of Chinese patients with genetic CJD who have E196A or E196K mutation in PRNP: comparative analysis of patients identified in the Chinese National CJD Surveillance System
- Evaluation of the impact of CSF prion RT-QuIC and amended criteria on the clinical diagnosis of Creutzfeldt-Jakob disease: a 10-year study in Italy
- MM2-type sporadic Creutzfeldt-Jakob disease: new diagnostic criteria for MM2-cortical type
- Systematic approach to diagnosing suspected Creutzfeldt-Jakob disease
- Imaging and CSF analyses effectively distinguish CJD from its mimics
- Intensity of human prion disease surveillance predicts observed disease incidence
- Comparison between plasma and cerebrospinal fluid biomarkers for the early diagnosis and association with survival in prion disease
- Molecular pathology of neurodegenerative diseases: principles and practice