Article Text

Abstract
Genetic and acquired disorders of white matter comprise a diverse group of conditions, with often overlapping clinical and radiological findings. Patients present with a variable combination of cognitive impairment, ataxia, spasticity or movement disorders, among others. There are many genetic causes, and the route to diagnosis involves comprehensive clinical assessment, radiological expertise, metabolic investigations and finally genetic studies. It is essential not to miss the treatable acquired causes. In this review, we present a practical approach to investigating patients with acquired and genetic disorders of white matter, based on the experience of a large international referral centre. We present a guide for clinicians, including pitfalls of testing, clinical pearls and where to seek advice.
- MULTIPLE SCLEROSIS
- GENETICS
- DEMENTIA
- CLINICAL NEUROLOGY
- MRI
- ALZHEIMERS DISEASE
- ADRENOLEUKODYSTROPHY
- NEUROGENETICS
- EPILEPSY
- METABOLIC DISEASE
- NEURORADIOLOGY
Statistics from Altmetric.com
- MULTIPLE SCLEROSIS
- GENETICS
- DEMENTIA
- CLINICAL NEUROLOGY
- MRI
- ALZHEIMERS DISEASE
- ADRENOLEUKODYSTROPHY
- NEUROGENETICS
- EPILEPSY
- METABOLIC DISEASE
- NEURORADIOLOGY
INTRODUCTION
Neurologists frequently encounter patients whose MR imaging shows white matter abnormalities. Such patients may present with various symptoms and signs, including cognitive deficits, movement and gait disorders, ataxia and many others. Occasionally, the finding is incidental, and the patient is apparently asymptomatic. Often the diagnosis is straightforward, and an acquired cause readily identified. However, there are some patients where there is no diagnosis despite extensive investigations. Many of these have a genetic disorder (ie, a leukodystrophy) and they require specialist input to advance their diagnosis.
In an effort to improve the care of these patients, we developed the Queen Square Adult Leukodystrophy Group, a multidisciplinary team of neurologists, radiologists and metabolic physicians who review the clinical presentations, investigations and neuroimaging of adults with white matter disorders throughout the UK and abroad.
Here, we describe our approach to these patients, with illustrative cases and tips on avoiding common pitfalls. It is not a comprehensive review, which may be found elsewhere1–3 but rather a framework to apply to such cases. Where specialist advice is needed, readers may contact various research groups or authors of this paper.
When to suspect acquired disease
The most common acquired causes that we see are multiple sclerosis and acquired small-vessel disease. Neurologists frequently encounter uncommon presentations of these common conditions, which raise the possibility of a leukodystrophy. These are important to recognise in order to consider specific treatments or risk factor management.
Neurologists are of course familiar with the diagnosis of multiple sclerosis—both the relapsing and progressive phenotypes—and know the well-described pitfalls.4 Difficulties can arise with atypical presentations, such as isolated cognitive involvement, or with end-stage disease, where the white matter changes may be confluent and somewhat symmetrical.
Re-assessment of the history may identify overlooked relapses, and a formal review of any previous imaging may identify earlier diagnostic clues. If uncertainty persists, then it can be very helpful to identify spinal involvement on MR imaging, unmatched CSF oligoclonal bands, and abnormal visual evoked potentials.
Beyond multiple sclerosis, other clues to an acquired cause include a relatively rapid onset and progression (significant deterioration within 6–12 months of onset), systemic features and MR imaging showing cranial enhancement or cervical cord involvement. Similarly, if patients have previously responded to corticosteroid treatment, an acquired cause is more likely.
In the history, ask about chemotherapy or radiotherapy exposure, as well as drug use. It is useful to enquire about symptoms that suggest inflammatory disorders, such as arthralgia, rashes, ocular symptoms and marked weight loss or fevers. A broad autoimmune screen should be performed, including testing for antinuclear antibody, antineutrophil cytoplasmic antibody, extractible nuclear antigen and the cardiolipin and lupus anticoagulant antibodies. A history of immunosuppression should prompt consideration of progressive multifocal leukoencephalopathy (also rarely occurring in the immunocompetent), and all patients should be tested for HIV, syphilis and hepatitis B and C and consideration given to tuberculosis. Finally, clinicians should consider neoplastic causes including gliomatosis and primary CNS lymphoma. Features strongly suggesting an acquired cause (inflammatory or neoplastic) include parenchymal swelling (in particular with gadolinium enhancement), certain patterns of diffusion restriction and rapid progression; in such cases, a brain biopsy might be considered.
We consider the investigation of acquired causes to be Round 1 investigations (table 1), the most critical step to avoid missing a treatable cause.
Recommended investigations in white matter disorders
Case 1
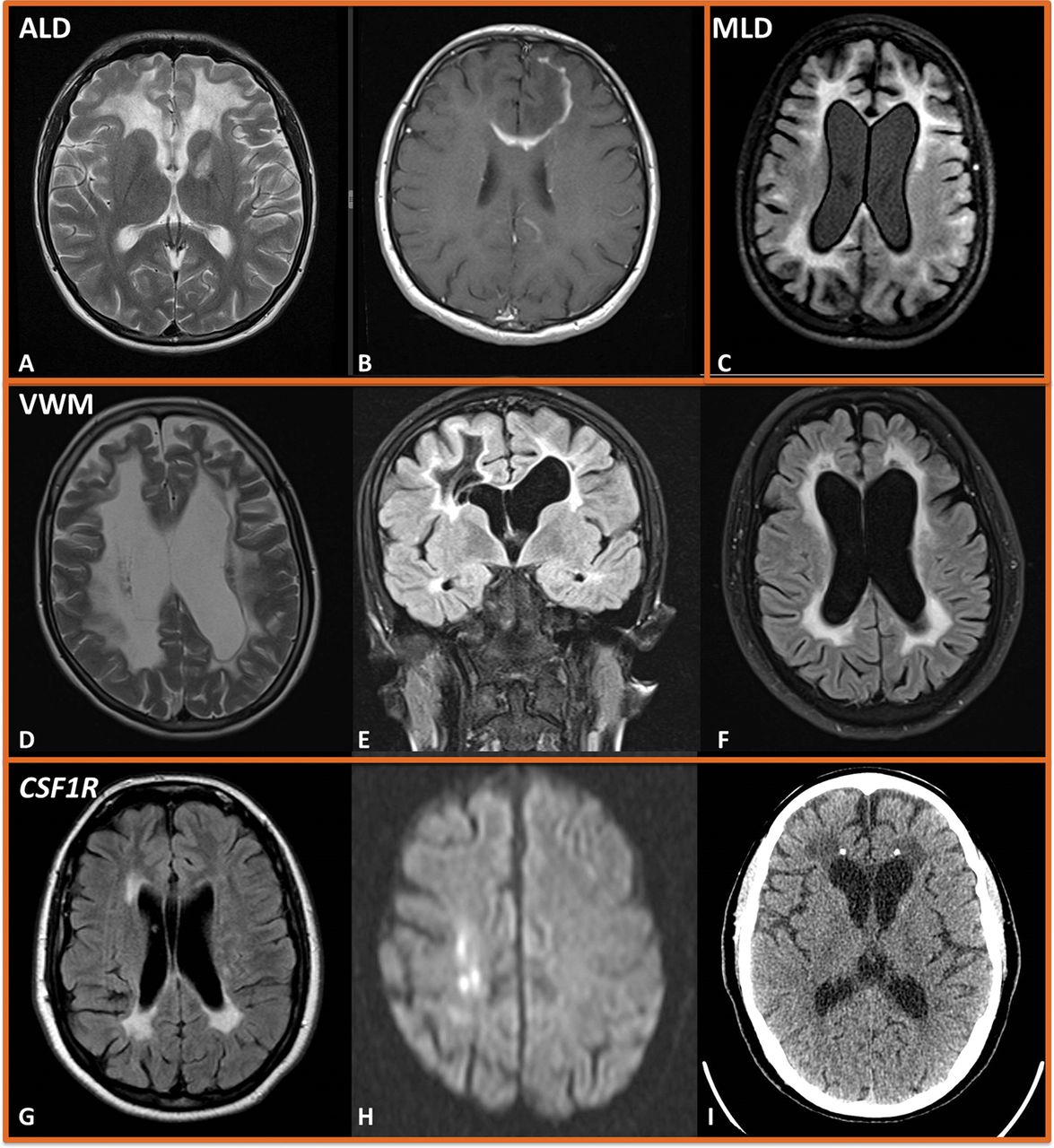
A 50-year-old woman was referred with a possible leukodystrophy. She had presented at age 40 with clumsiness of the right hand, and over the next 10 years had become increasingly unsteady, with subjective cognitive decline and bladder urgency. MR scan of the brain showed relatively symmetric and confluent signal change (figure 1A–B). However, there was a suggestion of multiple lesions that had coalesced, and there were distinct lesions in the cerebellum. Review of prior imaging at disease onset confirmed typical demyelinating lesions, and imaging of the spinal cord was recommended. STIR/T2-weighted imaging of the spine found typical short segment inflammatory lesions, and there were unmatched oligoclonal bands in the CSF (figure 1B). We diagnosed primary progressive multiple sclerosis with ongoing radiological activity and started ocrelizumab.

A–B: Primary progressive multiple sclerosis. Axial T2-weighted MR scan of the brain shows relatively symmetrical and confluent white matter signal change, but with periventricular multiple focal lesions that have coalesced (A). Sagittal T2-weighted MR scan of the brain shows short-segment inflammatory lesions in the cervical cord (B). C–D: HIV encephalopathy. Axial T2-weighted (C) and coronal FLAIR (D) MR scan of the brain showing symmetrical and confluent white matter high signal with involvement of the external capsule and anterior temporal lobes. E–G: Diffusely infiltrating glioma. Axial FLAIR (E) MR imaging showing asymmetrical frontal high signal with trans-callosal extension and cortical and subcortical swelling. There is an additional area of high signal posteriorly, and diffusion-weighted imaging (F–G) shows a frontal rim of restricted diffusion. H–J: Small-vessel disease. Axial T2-weighted (H) MR scan of brain showing patchy periventricular and deep white matter high signal, with involvement of the pons (I), basal ganglia and thalamus (J). FLAIR, fluid attenuated inversion recovery.
Case 2
A 65-year-old woman had been referred to the neurogenetic clinic after negative NOTCH3 testing. She was a retired nurse and had parasthesiae in both feet. Investigation identified a mild axonal neuropathy, lymphopenia and diffuse brain white matter changes on MR imaging, most likely representing small-vessel disease. Three years later, she had developed cognitive difficulties, as well as pyramidal signs in her legs. Her MR brain scan appearances had progressed significantly (figure 1C–D), with prominent involvement of the external capsules and anterior temporal lobes. CSF showed a raised protein of 1.39 g/L (0.15–0.45) with matched serum and CSF oligoclonal bands. Round 1 investigations identified her to be HIV positive. She improved clinically and radiologically with antiretroviral treatment.
Case 3
A 31-year-old woman had a background of mild intellectual impairment, longstanding unilateral deafness, short stature and diabetes. Nine months before her assessment, she had developed disinhibited behaviour. Five months later, she developed episodic vomiting, and over the next 3 months, she became more withdrawn with abnormal behaviour. She subsequently presented in status epilepticus, requiring intubation and intensive care unit support. She had frontal and pyramidal signs, and MR scan of the brain showed asymmetric bifrontal T2-weighted signal abnormalities with transcallosal extension, significant cortical and subcortical swelling and small foci of enhancement (figure 1E–G). CSF was acellular with raised protein (0.80 g/L (0.15–0.45)), matched CSF and serum oligoclonal bands and normal cytology.
Her background features and longer prodromal symptoms raised the possibility of a genetic disorder, but overall the rapid decline over 3 months and her imaging favoured an acquired cause. A frontal lobe biopsy showed a diffusely infiltrating glioma, WHO grade IV.
When to suspect severe small-vessel disease
The most common diagnosis (16%) reached by the Adult Leukodsytrophy Group on externally referred patients is severe acquired small-vessel disease.
Patients with acquired small-vessel disease typically have few symptoms or signs and may be asymptomatic. They are usually older (>60 years) with multiple vascular risk factors, for example, diabetes, hypertension, smoking, dyslipidaemia, renal disease. MR scan of the brain typically shows patchy T2-weighted/fluid attenuated inversion recovery (FLAIR) signal changes in the periventricular and deep white matter, which become confluent over time with involvement of the pons, thalamus and basal ganglia. Lacunar infarcts and deep microhaemorrhages support the diagnosis; spinal cord imaging is normal (figure 1H–J). In severe small-vessel disease, there may be involvement of the internal and external capsules, as well as the anterior temporal lobes, reinforcing that anterior temporal lobe signal change is not pathognomonic of monogenic small-vessel diseases (eg, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Conversely, when a patient is young (eg, younger than 40 years) with no typical vascular risk factors and/or with a suggestive family history, clinicians should consider a monogenic small-vessel disease (Case 4).
Case 4
A 32-year-old woman born of consanguineous parents gave a 3-year history of slowly progressive gait disturbance, headaches and subcortical cognitive dysfunction. MR scan of the brain showed patchy T2-weighted periventricular high signal and mature striatocapsular and pontine infarcts with scattered microhaemorrhages in the posterior fossa, thalami and basal ganglia. There was subtle anterior temporal lobe involvement.
The imaging was in keeping with a vascular leukoencephalopathy. Given the young age of onset and absence of vascular risks, we suspected a monogenic small-vessel disease. Single-gene testing for NOTCH3 had already been undertaken and was negative. Single-gene sequencing identified a homozygous loss of function mutation in HTRA1, confirming a diagnosis of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL).
How we interpret metabolic tests
Once acquired causes have been excluded, we request metabolic investigations in all patients (Round 2 investigations—Table 1). This includes very-long-chain fatty acids (X linked adrenoleukodystrophy/adrenomyeloneuropathy); white cell enzyme activity (specifically galactocerebrosidase (Krabbe disease) and arylsulfatase A (metachromatic leukodystrophy)); plasma amino acid profile and total homocysteine (homocystinuria and methylenetetrahydrofolate reductase deficiency); plasma sterols and urinary bile alcohols (cerebrotendinous xanthomatosis). These tests are relatively inexpensive (~£200) and accessible and the results can currently be obtained much more quickly than genetic panels. Together, they exclude >90% of all metabolic leukoencephalopathies that occur in adults and can be invaluable in interpreting variants identified by genetic testing.
Unless experienced in requesting these tests, we recommend contacting a metabolic laboratory in advance to ensure appropriate sampling and handling, as spurious results can be obtained if guidance is not followed. In particular, delays in sample processing will lead to falsely low results of enzyme activity, and difficulties in interpreting amino acid profiles. These tests, therefore, require planning and coordination between the neurologist, patient and laboratory to ensure valid results, and careful interpretation to avoid misdiagnosis.
Case 5
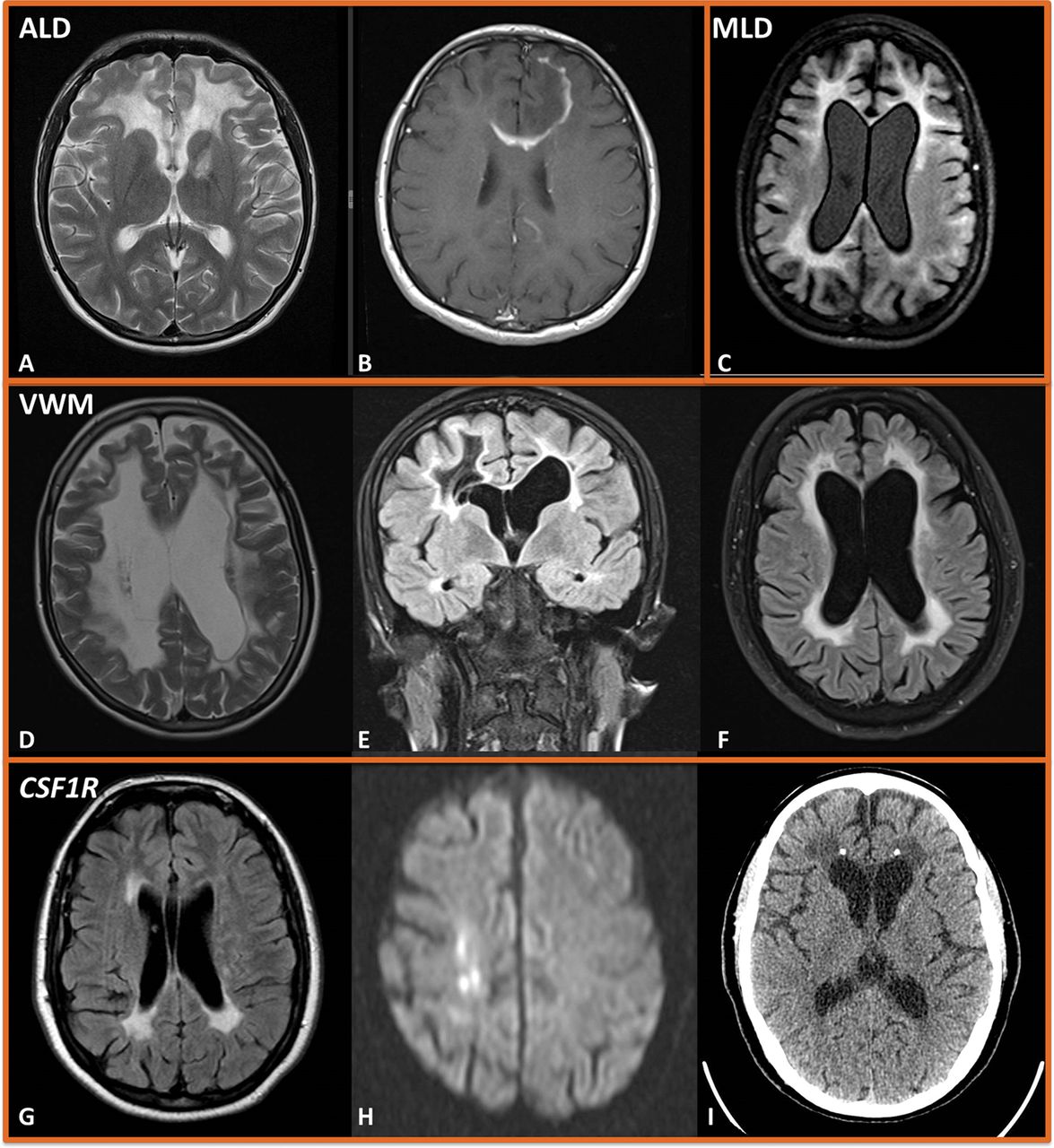
A 28-year-old man was referred following an abnormal MR brain scan. He had a background of adrenal failure diagnosed at 6 years of age, and mild subcortical dysfunction on cognitive testing. T2-weighted imaging showed confluent and symmetrical increased signal in the subcortical white matter of the frontal lobes, sparing the U-fibres and extending into the genu of the corpus callosum. T1 postcontrast sequences showed enhancement in the left frontal lobe and genu (figure 2A–B). A very-long-chain fatty acid profile identified raised C24:C22 and C26:C22 ratios (table 2).
Very-long-chain fatty acid profile
{kind=link}
{kind=link}
A–B: Adrenoleukodystrophy. Axial T2-weighted (A) and T1-gadolinium-enhanced (B) MR scan of brain showing confluent and symmetrical white matter high signal in the frontal lobes, with enhancement in the left frontal lobe extending into the genu of the corpus callosum. C: Metachromatic leukodystrophy. Axial FLAIR (C) showing symmetrical and confluent high white matter signal, with frontal predominance and associated volume loss. D–F: Vanishing white matter disease. Axial (D) and coronal T2-w (E) and FLAIR (F) MR scan of the brain showing confluent and symmetrical white matter high signal with volume loss. T2-weighted FLAIR shows periventricular rarefaction, with the white matter developing the same signal as the CSF—a characteristic feature of vanishing white matter disease. G–I: CSF1R. Axial FLAIR (G) MR scan of the brain showing areas of periventricular high signal at the frontal and occipital horns. Diffusion-weighted imaging shows punctate areas of restriction in the deep white matter (H), and CT scan of head shows bilateral foci of calcification (I)—both characteristic of CSF1R (hereditary diffuse leukoencephalopathy with spheroids). FLAIR, fluid attenuated inversion recovery.
Targeted single-gene testing of ABCD1 identified a hemizygous missense variant in exon 1. We diagnosed X linked adrenoleukodystrophy/adrenomyeloneuropathy, with active cerebral demyelination based on the postcontrast enhancement. Most patients present with involvement of the splenium and parieto-occipital white matter, but this less common frontal form is well recognised. Allogenic haematopoietic stem cell transplantation may have a role in selected cases.5
Case 6
A 38-year-old woman was referred following a seizure and abnormal MR scan of the brain. She had normal development milestones and had been previously well. Her parents were consanguineous. The family reported a 4-year history of cognitive decline. She had stopped working due to this and subsequently had developed episodes of incontinence, focal seizures, increasingly child-like behaviour and disinhibition. MR scan of the brain showed symmetrical confluent periventricular T2-weighted signal abnormality with frontal predominance and volume loss (figure 2C). Nerve conduction studies identified a generalised demyelinating peripheral neuropathy. Arylsulfatase A activity was very low (4 nmol/hour/mg, normal range 22–103), and targeted single-gene sequencing confirmed a diagnosis of metachromatic leukodystrophy.
How we do genetic testing
We proceed to genetic testing only after reviewing the results of Round 1 and 2 investigations. The potential implications should be discussed with the patient and their family in advance. We interrogate the phenotype both clinically and radiologically, to see if we can identify a likely single gene, or group of disorders (eg, genes causing hypomyelination). Although the clinical and radiological appearances overlap between many leukodystrophies, there are useful signs that we use to guide our testing. Clinically, finding endocrine abnormalities is very useful, in particular adrenal failure (X linked adrenoleukodystrophy/adrenomyeloneuropathy), ovarian failure (vanishing white matter disease, AARS2-related leukodystrophy) or hypogonadism (Gordon Holmes syndrome). Parkinsonism may suggest mutation in the CSF1R gene, or ataxia may suggest a mutation in the CLCN2 gene. Neuropathy may be either demyelinating (metachromatic leukodystrophy) or axonal (X linked adrenoleukodystrophy/adrenomyeloneuropathy or cerebrotendinous xanthomatosis).
Vanishing white matter disease (figure 2D–F), CSF1R (figure 2 G–I) and AARS2-related leukodystrophies have relatively specific imaging appearances. Specific signs, such as hypomyelination or prominent posterior fossa abnormalities, may suggest groups of disorders. Our recent review in JNNP provides a more comprehensive guide to clinical genetic correlation.2
Single-gene testing remains important for confirming a metabolic leukodystrophy suspected from the presentation and Round 2 investigations. However, beyond this role, and despite rigorous phenotyping, we no longer recommend single-gene sequencing. Genetic panel testing can usually be performed with a similar cost and turnaround time, with the advantage of sequencing multiple genes simultaneously, which is useful in heterogeneous disorders. Genetic laboratories usually use a generic sequencing panel that sequences thousands of genes but restrict their analysis to relevant genes. This is to prevent the clinician from being overwhelmed by variants that are not relevant to the patient’s presentation. Genetic panels have their limitations, and it is important to be aware of these (box 1).
Tips and pitfalls of genetic testing
Genetic testing
Single-gene testing is useful to confirm a metabolic disorder suspected from Round 2 investigations, but beyond this, a leukodystrophy panel is the most efficient route to diagnosis
Be aware that routine panels do not identify some types of mutation (eg, large deletions, duplications and expansions); thus, a negative panel does not rule out a gene completely. Correlate with the clinical phenotype and if there is a strong suspicion despite a negative genetic panel, contact a research group
Different genetic laboratories construct their own unique panels, so the list of genes sequenced varies, depending on where the test is done. It is important to request a broad ‘leukodystrophy/leukoencephalopathy’ or ‘white matter disorders’ panel. The full list of targeted genes will be available from the laboratory
Traditional panels do not capture mutations in mitochondrial DNA (mtDNA), and these are not detected well in DNA extracted from blood. Consider a muscle biopsy with mtDNA sequencing from muscle if there are suggestive features.
Whole- exome or genome sequencing may give a higher yield, although expertise is needed in interpretation
Because panels sequence many genes simultaneously, many variants are identified. Most are harmless polymorphisms, not relevant to the patient or their disorder. It can be difficult to determine which variants are pathogenic and which are of no clinical significance. Variants that cause rare diseases should themselves be exceedingly rare in the population, and usually lead to a change in protein function or expression. Very often, the laboratory reports ‘variants of uncertain significance’ where there is not enough evidence from the literature, population databases or prediction tools to decide if the variant is benign or pathogenic. These require experience to interpret, in particular to determine whether the genotype matches the phenotype radiologically and clinically (box 2).
Tips and pitfalls in metabolic testing
Pitfalls in metabolic testing
Time delay between the taking of samples to processing will lead to falsely low activity levels of most enzymes. To ensure appropriate samples, storage and timely transport, discuss with the laboratory first and coordinate the best time and place for sampling.
Hyperlipidaemia can increase very-long-chain fatty acids. A repeat fasting sample taken together with a lipid profile may be needed to aid interpretation.
Pseudodeficiency alleles
Pseudo-deficiency alleles involve a variant in the gene encoding the enzyme that leads to low enzyme activity levels, but not low enough to cause the disease.
They are particularly common in the ARSA gene (arylsulfatase A, metachromatic leukodystrophy) where they are carried by ~2% of the population.
If low enzyme activity is reported despite appropriate sample handling, pseudodeficiency alleles should be considered. Discuss with the laboratory and consider confirmation with genetic analysis
How to interpret heterozygous variants in recessive genes
Mutations on both alleles are required to cause recessive disease. When a heterozygous variant is found, consider either:
The phenotype matches the variant found: there is likely a second pathogenic variant not detected (eg, a large deletion or intronic variant that is not captured by the panel used). Consider specialist opinion and research groups
The phenotype does not match the variant found: the patient is likely a carrier of the variant and it is of no relevance to the presentation. Beware very rare variants known to cause disease. Do you need to re-evaluate the phenotype?
Case 7
A 45-year-old woman was referred with cognitive decline. She had no significant previous medical or family history. She had a 2-year history of increasing difficulty with tasks at work, culminating in her being made redundant. She had subsequently developed episodes of incontinence, had difficulty following conversations and latterly had required assistance in activities of daily living. She had marked frontal and subcortical cognitive deficits, a parkinsonian gait and broken saccades.
Nerve conduction studies were normal. MR scan of the brain showed symmetrical periventricular T2-weighted signal abnormality with a frontal predominance and volume loss. The external capsule and temporal poles were spared, and there were no microhaemorrhages. Arylsulfatase A activity was moderately reduced, but not as low as in metachromatic leukodystrophy. Following discussion with the laboratory, genetic analysis confirmed the presence of an arylsulfatase A pseudodeficiency allele, a common and harmless finding. NOTCH3 sequencing identified a heterozygous mutation, previously reported as pathogenic. The patient and family were informed of the diagnosis of CADASIL. Following reassessment of the phenotype and genotype, the NOTCH3 variant was found to be a rare polymorphism, unlikely to cause the phenotype, which was also not typical for a monogenic small-vessel disease. Further genetic analysis revealed a pathogenic variant in the CSF1R gene, confirming a diagnosis of hereditary diffuse leukoencephalopathy with axonal spheroids.
CONCLUSIONS
General neurologists commonly encounter patients who have prominent white matter changes on MR brain imaging, and usually the diagnosis is straightforward. However, when a diagnosis is not apparent, there is often uncertainty as to how best to investigate, and particularly when access to specialist tests; expert clinical and radiological opinions may not be readily available.
In such cases, we suggest following a structured approach:
Round 1 tests involve a detailed assessment for acquired causes. Further neuroimaging (including longitudinal and retrospective review of prior images when available) of both brain and cervical spine with gadolinium can be particularly helpful, as can CSF examination.
Round 2 tests for metabolic disorders should be performed following discussion with a metabolic laboratory and carefully planned to ensure appropriate sampling, handling and transport.
Round 3 is where a genetic diagnosis is considered. It is usually most efficient to request genetic panel testing, which is becoming more widely available. Clinicians should be aware of the high rate of incidental findings, the potential limitations of genetic panels and the difficulties interpreting heterozygous variants in recessive disorders, and consider whether any genetic results fit with the phenotype. For this reason, we advocate seeking specialist advice once genetic testing is considered. This may be through a regional neurogenetics or clinical genetic service.
Clinicians are welcome to refer cases to be reviewed remotely or in person by the Queen Square Adult Leukodystrophy Group.
Key points
The priority for investigating a white matter disorder is to exclude treatable and acquired causes; clinicians should thoroughly investigate for infections, neoplasia and immune disorders (Round 1) before requesting any metabolic and genetic investigations.
Take care when requesting and interpreting metabolic investigations (Round 2), and note that pseudodeficiency states are common and do not cause disease; if in doubt, discuss handling with the laboratory.
Certain clinical and radiological signs help to narrow down the likely genetic cause; some features, such as calcification on CT scan of brain or early ovarian failure, may need to be specifically sought (Round 3).
Footnotes
Contributors TW, HH, EM, NJ, NCF, JMS, MA, ID, JC and DSL authors participated in drafting the manuscript and figures. Final oversight and approval provided by JC and DSL.
Funding MRC, Wellcome Trust, UCLH Biomedical Research Centre.
Competing interests None declared.
Ethical approval Not required.
Provenance and peer review Commissioned. Externally peer reviewed by Emma Tallantyre, Cardiff, UK.
Other content recommended for you
- A practical approach to diagnosing adult onset leukodystrophies
- Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era
- Diffusion and ADC-map images detect ongoing demyelination on subcortical white matter in an adult metachromatic leukodystrophy patient with autoimmune Hashimoto thyroiditis
- Encephalopathy in a 45-year-old woman: presented at the Advanced Clinical Neurology Course, Edinburgh 2010
- Myelopathy but normal MRI: where next?
- Adrenoleukodystrophy: a forgotten diagnosis in children with primary Addison's disease
- Fifteen-minute consultation: A practical approach to developmental regression in children
- Peripheral neuropathy in complex inherited diseases: an approach to diagnosis
- Imaging in multiple sclerosis and related disorders
- Adult-onset adrenoleukodystrophy presenting with status epilepticus and psychosis