Article Text

Abstract
Delirium is an acute disorder of fluctuating attention and awareness with cardinal features that allow it to be positively distinguished from other causes of an acute confusional state. These features include fluctuations, prominent inattentiveness with other cognitive deficits, a change in awareness and visual hallucinations. We describe a framework for diagnosing delirium, noting the need to consider certain caveats and differential diagnoses. Delirium is a clinical diagnosis where a thorough history and clinical examination are much more helpful diagnostically than any single test or combination of tests.
- GERIATRICS
- COGNITION
Data availability statement
No data are available.
Statistics from Altmetric.com
Introduction
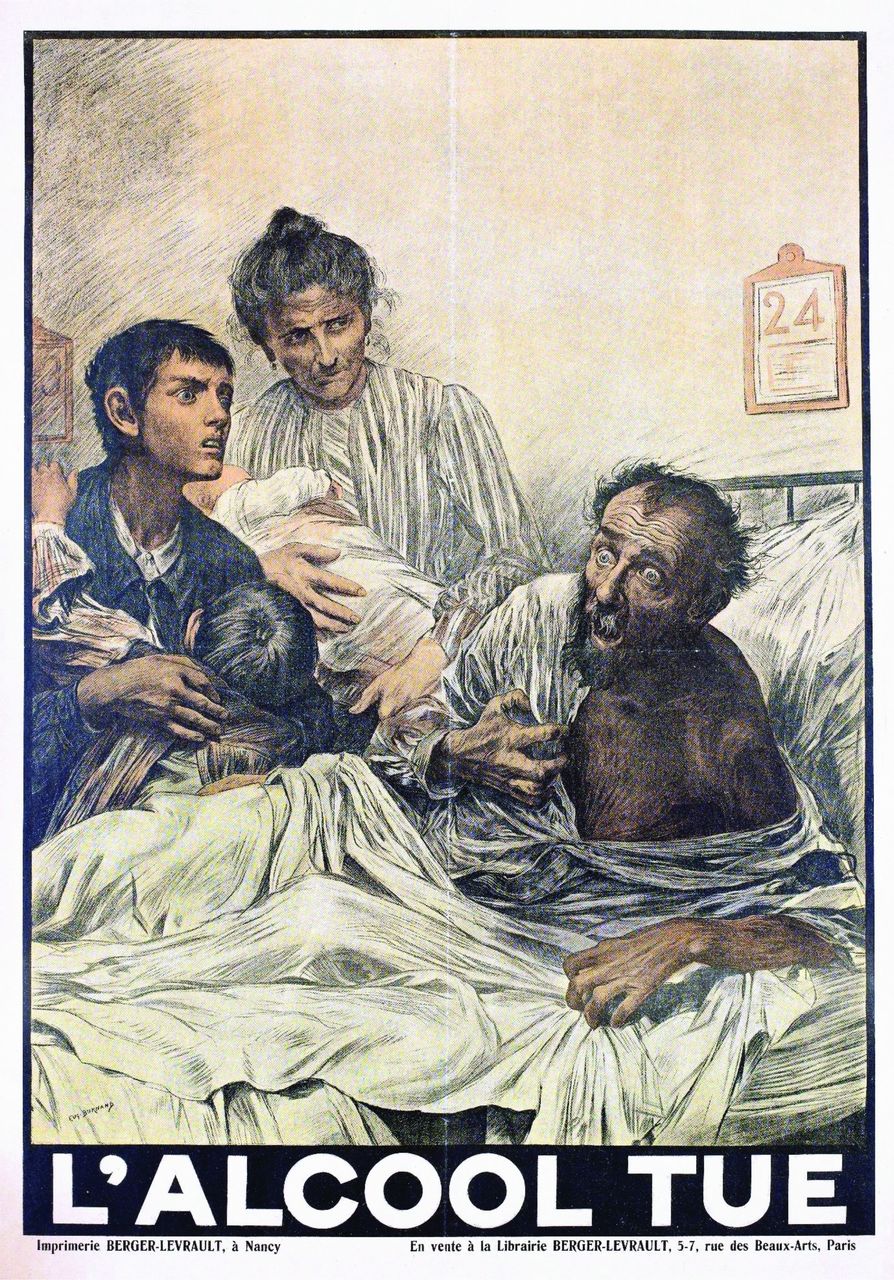
Delirium is an acute disorder of fluctuating attention and awareness. It is very common among hospital inpatients (one third of >65 years in some studies) and is mostly synonymous with septic or metabolic encephalopathy.1 Box 1 summarises its different manifestations and figure 1 shows the historical poster illustrating some features of hyperactive delirium.2–4
Criteria for the diagnosis of delirium2–4
Delirium is defined in DSM-V as:
Disturbance in attention (ie, reduced ability to direct, focus, sustain and shift attention) and awareness (reduced orientation to the environment).
The disturbance develops over a short period of time (usually hours to a few days), represents an acute change from baseline attention and awareness, and tends to fluctuate in severity during the course of a day.
An additional disturbance in cognition (eg, memory deficit, disorientation, language, visuospatial ability or perception).
The disturbances in criteria A and C are not better explained by a pre-existing, established or evolving neurocognitive disorder and do not occur in the context of a severely reduced level of arousal such as coma.
There is evidence from the history, physical examination or laboratory findings that the disturbance is a direct physiological consequence of another medical condition, substance intoxication or withdrawal (ie, due to a drug of abuse or to a medication), or exposure to a toxin, or is due to multiple causes.
There are four different types of delirium:
Hyperactive delirium is characterised by motor agitation, restlessness and sometimes aggression.
Hypoactive delirium is the most common form of delirium, characterised by motor retardation, apathy, slowness of speech and appearance of being sedated.
Mixed delirium has components of hyperactive and hypoactive delirium.
Delirium without motor features (cognitive symptoms only).

The wide-eyed stare of this man with delirium, in this case due to alcohol, features in this poster from the early twentieth century ‘alcohol kills’. Taken from Wellcome Collection under Creative Commons license, accessed 27 August 2022.
While the term ‘delirium’ is useful for communicating between clinicians a recognisable clinical entity, it is widely encompassing and includes multiple clinical syndromes and underlying pathophysiological entities, rather than a standalone diagnosis. Indeed, alternative terms such as encephalopathy or even ‘brain failure’ may be preferable, as they highlight the analogy with other organ failures (eg, heart failure, renal failure) where clinicians recognise the relative contributions of both pre-existing organ health and acute insult to a clinical decompensation (figure 2). This conceptual framework emphasises that ‘delirium’ is the start of a diagnostic formulation rather than a full explanation (‘delirium secondary to…’ rather than ‘delirium.’). The diagnosis should prompt further questions: Why this patient? Why now?

{kind=link}
{kind=link}
The interaction between severity of physiological insult and brain vulnerability. mRS, modified Rankin scale.
Delirium is not a diagnosis of exclusion: it has its own cardinal features of fluctuations, prominent inattentiveness with other cognitive deficits, changes in awareness and visual hallucinations, and temporal association with a provoking trigger (and improvement with treatment or removal of that trigger). Here, we describe a framework for its diagnosis and note particular caveats and differential diagnoses to consider. We emphasise that delirium is a clinical diagnosis where thoroughness of history-taking and clinical examination are diagnostically much more helpful than any single or combination of paraclinical test(s). Delirium is not the only cause of an acute confusional syndrome: although statistically likely in an older person with pre-existing cognitive or sensory impairments, it important to identify alternative and treatable neurological conditions that may mimic delirium. As with other diagnostic labels, a hasty diagnosis of delirium can curtail diagnostic thinking.
The differential diagnosis of the acutely confused patient has expanded in the past 10 years to include autoimmune and medication-related causes in particular; however, its diagnosis and management is largely unchanged. In this paper, we focus on its diagnosis and acknowledge sources of helpful information that discuss its prevention (up to 30% delirium is preventable) and management (mostly non-pharmacological, but antipsychotics where required are safe and do not prolong delirium).5–9
Pathophysiology of delirium
The pathophysiology of delirium remains uncertain, partly because of the diverse clinical states collected under this umbrella term. For example, is the aggressive agitation in a 24-year-old on Intensive Care Unit (ITU) the same condition as the obtundation of a 98-year-old with Alzheimer’s disease with a relatively minor urinary tract infection? The probability of developing delirium is clearly driven by the combination of underlying cerebral vulnerability and the magnitude of systemic upset (see figure 2).
There are several proposed mechanisms but (despite having been postulated for decades) none has a convincing evidence base and none is likely to explain the whole clinical syndrome in isolation.
Neurochemical imbalance
A leading hypothesis is relative cholinergic deficit, which has face-validity to clinicians aware of the detrimental cognitive effects of anticholinergic medications and the benefits of cholinergic augmentation in Alzheimer’s disease. Positron emission tomography and single-photon emission CT studies have shown perfusion abnormalities that co-localise with central cholinergic pathways in delirium and experimental pharmacological reduction of acetylcholine can, in isolation, precipitate a delirium state.10 11 However, there is little further experimental support for this hypothesis, and therapeutic augmentation of acetylcholine does not seem to help.12 Dopaminergic antagonists are commonly used to manage challenging agitation in delirious patients, but beyond the obvious sedative effects, there is no evidence that they change the underlying pathophysiology.13–15
Neuroinflammation
Conditions that induce peripheral inflammatory responses are common triggers for delirium, and peripheral inflammation also causes neuroinflammation. Intravenous injection of lipopolysaccharide, an experimental model of Gram-negative sepsis, leads to microglial activation in humans, a phenomenon supported by postmortem findings in those who have died from sepsis.16 17 Mediators secreted by activated microglia can have direct deleterious effects on neuronal function, as well as indirect effects on neuronal metabolism.18 Similarly, there may be elevated intrathecal concentrations of series proinflammatory mediators (such as IL-1b and IL-8) in patients with delirium, although it is challenging to interpret these data in the face of permeabilised blood–brain barrier (another potential contributor to delirium).19 20
Brain energy metabolism
A failure to maintain adequate brain energy supply during states of acute physiological stress has long been a possible explanation.21 Clearly, frank deficiencies in metabolic substrates such as glucose or oxygen can impair brain function, but the relevance to delirium more broadly is unclear; cerebral autoregulation does appear to be impaired in delirium secondary to sepsis, which may affect flow-metabolism coupling.22 Cerebrospinal fluid (CSF) lactate concentrations are elevated, which may represent either a deficiency of substrate, or a failure of its effective use by mitochondria.23 24
Relevance to COVID-19
There has been a surge of interest in delirium related to the prevalence of delirium in people with, and recovering from, COVID-19.25 To date, this interest has not delivered a paraclinical test that can positively identify individuals with, or at risk of, delirium, and this remains a focus for ongoing research.
What is the significance of a diagnosis of delirium?
Delirium reflects the interplay of the acute insult(s) to homeostasis with the degree of brain frailty (figure 2). As delirium is a temporary condition associated with a provoking factor, and commonly causes significant stress and anxiety, it is tempting to be reassuring to the patient and family. However, delirium is associated with an increased risk of future dementia and mortality (5–11 fold increase in risk of dementia in several studies).26 Following up a diagnosis of delirium with counselling about lifestyle changes can delay the onset of dementia and discussion of practical matters such as lasting power of attorney can offer potential (but unproven) benefit.
One-third of cases of delirium may be avoidable, and so it is worth taking sensible steps to reduce its risk. It is not yet known whether delirium itself causes permanent neuronal damage.5 6
Common mimics of delirium: how to recognise them
Neurologists are often asked of patients with delirium, ‘Is this something else; Are we missing something?’ Neurologists rarely make the initial assessment of delirium but typically become involved a few days later when the patient has not improved with treatment or where no there is no identifiable systemic infection, metabolic disturbance or pharmacological precipitant. However, by this time, the patient’s clinical condition has often been compounded by their admission to an unfamiliar hospital environment.
The cornerstone to management starts with a structured and thorough approach to information gathering. Context is important in the formulation of delirium and its mimics, and so obtaining collateral information from friends, family and neighbours is key. Additionally,
General practice records may describe recent symptoms, new medications and previously diagnosed risk factors for delirium.
Ambulance transfer documents may describe the situation at home—particularly helpful with patients living alone—including signs of self-neglect or alcohol misuse, or any pointers to seizures, such as loss of urinary or faecal control, hypoglycaemia, hypoxia or head injury at the scene. The condition of the patient during transfer to hospital is also important.
Nursing records may detail sleep–wake cycle disturbance and provide evidence of fluctuating cognition, adding confidence to the diagnosis of delirium.
The duration and location of hospital care may be relevant.
Unsurprisingly, the strongest predictors of delirium are intensive care admission and prior dementia. However, other studies have identified that the next most predictive factors include pressure ulcers (at any stage of admission), non-elective admission, serum creatinine >133 µmol/L and a fracture of any type; all these are more predictive than age alone.27 Figure 2 shows the interplay between the acute insult and brain frailty.
Neurological examination is focused on identifying the key diagnostic features of delirium and identifying localising signs that might indicate more than generalised brain dysfunction. A particular challenge to its diagnosis is that each of the cardinal features is not specific, and each feature in isolation has a smaller differential diagnosis (see table 1). Identifying fluctuating cognition and behaviour is central to diagnosis, so it is valuable to return to and repeat parts of the assessment.
Key features of delirium and differential diagnoses to consider
Box 2 summarises a proposed hierarchy of investigations. First-line investigations, appropriate for almost everyone with delirium, aim to identify an underlying cause and so to guide specific treatment. Second-line, third-line and fourth-line investigations are tailored to the individual case, looking for a non-delirium diagnosis. These are most relevant when there are few markers of cerebral frailty, a small (or no) clear acute insult and/or where there are unexplained clinical features atypical for delirium. Patients in whom there is only limited or unavailable collateral history often require more investigations.
Hierarchy of investigations
First line: for most people with delirium
Basic observations including oximetry.
Full blood count, urea and electrolytes, liver function tests, thyroid stimulating hormone, adjusted calcium.
Infective screen: urine microscopy, culture and sensitivity (not dipstick), chest X-ray, COVID-19 swab, blood cultures.
Blood gas (venous sample usually sufficient to exclude carbon monoxide toxicity and carbon dioxide retention; arterial sample appropriate if narcosis is suspected).
Second line: low threshold for these additional tests when delirium remains likely
CT scan of head.
HIV, syphilis.
Serum B12.
Toxicology (urine drugs of abuse screen±blood alcohol concentration).
Serum ammonia (if abnormal liver function or taking sodium valproate).
Focused microbiological investigations.
Third line: Looking for specific neurological causes because either the context does not suggest a delirium OR delirium is unlikely from the acute insult+evidence of brain frailty
CSF (glucose, protein, lactate, cell count, viral PCR, consider store sample).
Electroencephalogram (EEG).
MR scan of brain.
Specialised blood tests
Autoimmune panel: Lgi-1, Caspr2 and N-methyl-D-aspartate (NMDA) receptor antibodies, thyroid peroxidase antibodies.
Paraneoplastic/antineuronal antibodies.
C3/C4, antinuclear antibodies, erythrocyte sedimentation rate.
Urinary porphyrins.
Fourth line: specialised tests for selected cases
Alzheimer’s disease CSF biomarkers, further serum antibodies.
Brain CT positron emission tomography scan (if suspecting degenerative illness).
Where the clinical assessment cannot establish a diagnosis of delirium, patients require more detailed investigations to exclude potential mimics. The extent to which more investigations are pursued depends on the clinical suspicion of an alternative diagnosis: this is the Gestalt of the assessment. The framework presented here emphasises the primacy of the clinical diagnosis ahead of rarefied tests. Table 2 lists the red flags that raise suspicion of an alternative (or coexistent), non-delirium diagnosis.
Red flags: clinical findings suggesting a diagnosis other than delirium or where a higher index of suspicion for alternative diagnoses is appropriate
Note that the specific clinical features of recently recognised autoimmune encephalitides clearly distinguish these from each other and from other causes of an acute confusional state, including delirium.28
Challenging scenarios: case examples
The neurologist’s assessment can be particularly valuable in assessing a potential delirium diagnosis in several situations. Here, we focus on three illustrative cases.
Case 1: a patient on the intensive care unit
A 32-year-old woman on the intensive care unit has not woken following a renal transplant for polycystic kidney disease. Her postoperative period was complicated by severe sepsis from an infected perinephric collection that was controlled only after radiologically guided drainage; her sepsis led to acute respiratory distress syndrome, and only now (2 weeks later) can her sedation be stopped. Despite 2 days off sedation, she has not woken, and shows little movement other than scanty myoclonic jerks noted during nursing care. Physiologically, she is otherwise stable; her gas exchange is good, and her unsupported blood pressure is 175/95 mmHg. Her renal function is deranged but stable without haemofiltration, with a urea of 24 mmol/L (2.5–7.8) and good urine output. Her liver injury and synthetic function tests are also deranged but improving. There is no active infection. She is off all medications other than tacrolimus (serum concentrations in range) and prednisolone. On examination, her eyes rest in the primary position but with a full range of conjugate movement on passive head movement and normal pupillary light and corneal reflexes. She spontaneously triggers the ventilator, and coughs on deep suctioning. She grimaces and mounts a tachycardia to pain but makes little movement of her limbs. Her reflexes are symmetrically brisk, and her plantars mute.
The duration of this patient’s intensive care stay and multiorgan failure is relevant, as many sedative and analgesic drugs (such as midazolam and morphine) accumulate given prolonged use, particularly in the setting of renal and hepatic dysfunction. Barbiturates are particularly difficult, as they accumulate in adipose tissue over time, and can subsequently leach back into the blood for a long period after infusions have ceased. Newer agents such as propofol and fentanyl tend to accumulate less, but are not exempt. Furthermore, this patient’s persistent moderate uraemia, which in isolation would be unlikely to cause such a profound encephalopathy in a young person, can, in the setting of other metabolic perturbations and a permeabilised blood–brain barrier, contribute powerfully to encephalopathy. Therefore, the most likely reason she has not woken appropriately is simply the culmination of multiple adverse metabolic/medication effects.
Myoclonus raises the possibility of underlying status epilepticus, and obtaining an EEG in this setting is important, as electrographic seizures can be remarkably common in severe ‘non-neurological’ illness. However, myoclonus is of course also common in all causes of diffuse cortical dysfunction such as metabolic encephalopathy.29 30
Non-convulsive seizures can be difficult to identify outside of an ITU setting. It is more likely after a generalised tonic-clonic seizure, in a patient known to have epilepsy or brain injury, and in women. EEG is better than clinical assessment in exploring this cause for acute confusion.31 However, EEG is sometimes practically difficult if there is no direct access to neurophysiology. There is ongoing research into alternatives to standard EEGs, including simpler recording technology and artificial intelligence-led EEG interpretation.32 In patients with acute confusion of uncertain cause who have risk factors for seizures, clinicians should have a moderately low threshold for obtaining an EEG, or for giving an empirical trial of antiseizure medication.
Relative hypertension in a patient taking tacrolimus raises suspicion of posterior reversible encephalopathy syndrome (PRES). While hypertension per se is unusual in intensive care (although not so unusual in neurocritical care, where both dysautonomia and therapeutic iatrogenic hypertension are relatively commonplace), medications, such as tacrolimus, occasionally cause PRES. An MR scan of the brain is therefore usually appropriate in this setting, although we recognise that transporting a critically ill patient for a long scan may have risk. Pathologies with important treatment or prognostic implications such as PRES, watershed or vaso-occlusive ischaemia, hypoxic brain injury, and fat emboli can be diagnosed on imaging, as can more obscure processes such as critical-illness-associated microbleeds and cytotoxic lesions of the corpus callosum.
The critical care unit setting substantially increases her risk of delirium, but her risk of iatrogenic and secondary brain injuries is also increased; all these factors may influence prognosis. Thus, it is often difficult to avoid investigations in this context.
Case 2: a haematology patient
A 66-year-old man received an allograft bone marrow transplant for chronic myeloid leukaemia 40 days ago. He has been recovering well, and until last week was self-caring and mobile without aids around his room. He has hypertension, chronic kidney disease (stage III) and distant history of obstructive sleep apnoea, (but last needed continuous positive airways pressure, CPAP 10 years ago). He takes tacrolimus for immunosuppression. In the past week, nursing staff have found him more sleepy than usual. Yesterday, he required assistance to dress and today, he fell beside his bed with urinary incontinence. His blood counts are incrementing as expected, his renal and liver function tests are all stable, and serum tacrolimus concentrations are consistently in range. On examination, he is disorientated in time and place and struggles to engage long enough to perform any bedside cognitive testing. He has no focal cranial nerve or limb signs.
This patient is early in his recovery and heavily immunosuppressed. He has non-specific features of an encephalopathy where drowsiness is the principal problem, but the fall and loss of continence could have been an unwitnessed seizure and support an encephalitic illness.
He is at risk of CNS and systemic infection, which may manifest without a clear systemic inflammatory response, and even CSF can be falsely reassuring in this setting. In addition to the usual concern about viral encephalitis and bacterial meningoencephalitis, human herpesvirus-6 (HHV-6) may present with an amnestic syndrome early after stem cell transplantation. An MR brain scan can show hyperintense mesial temporal lobe changes, but HHV-6 polymerase chain reaction (PCR) on CSF would be diagnostic. CNS infection with cytomegalovirus, toxoplasma and fungi are also possible concerns, justifying a low threshold for further investigation.
He is also vulnerable to medication toxicity and clinicians need to be familiar with such treatment specific side effects.33 Haematology patients are often exposed to highly potent (and CNS toxic) drugs, including methotrexate, and recently CAR-T cell therapy. He is also taking tacrolimus, which can cause symptoms like this (on a spectrum that includes PRES, and even if serum concentrations are within the therapeutic range). We have seen patients with methotrexate encephalopathy where imaging features became apparent only several weeks after initially non-specific clinical features. Likewise, CNS neurotoxicity (I-CANS) due to cytokine release syndrome induced by novel CAR-T cell therapy is often associated with few focal signs at early to moderate stages, and has specific treatments.
However, despite the detailed CNS tests, the answer lay in the history: his distant history of obstructive sleep apnoea was most relevant: repeat arterial sampling showed elevated carbon dioxide concentrations suggesting the sleep apnoea had decompensated. His wakefulness improved with CPAP, and within a week he was performing well on bedside cognitive testing. Detailed investigations for a coexistent neurological explanation were normal, including CT and then MR scans of brain, CSF examination including negative HHV-6 PCR, and EEG showed non-specific slowing without epileptiform features. An earlier trial of CPAP could potentially have avoided the need for such detailed investigation.
Case 3: a psychiatric patient
A 45-year-old man is brought to hospital by his ex-wife. He lives alone, but they see each other regularly. She describes him behaving increasingly oddly in the past 2 weeks. He has been reluctant to speak to his wife on the phone or in person, and appears irritable and distracted, often getting up to walk off halfway through a conversation. He has a history of alcohol excess, but has been teetotal for 2 years. He does not use recreational drugs. He is prescribed ramipril and sertraline.
On examination, he is disoriented to time and place. There is no aphasia but is perseverative. His neurological examination is otherwise normal with no cortical signs or features of meningism. He appears agitated and frequently paces up and down the ward. He appears uncomfortable and fidgets but there are no automatisms. He repeatedly asks why he is here and asks the doctor who they are. The nursing staff report that this behaviour is consistent day and night. His sleep is poor, and he is agitated overnight.
First-line and second-line blood tests were normal (see box 2). Urine toxicology was negative for drugs of abuse. Urinary porphyrins were negative. NMDA antibodies on CSF and serum were negative, LGI-1 and CASPR-2 antibodies were also normal. CSF amyloid-b and tau concentrations were normal. MR scan of brain was also normal.
He was managed for suspected acute alcohol withdrawal because of previous alcohol misuse. Although this was never proven with alcohol blood concentrations, it remained of concern as a potential cause of his acute deterioration. Despite extensive testing, it was only with the passage of time, when evidence of paranoid thinking and auditory hallucinations became clearer, that his diagnosis became clearer and he was diagnosed with an underlying psychotic disorder.
The case illustrates the challenge of how far to investigate for neurological disorders in a first episode of psychosis. In older patients, first episode psychosis may be the first presentation of Alzheimer’s disease.34 In younger people, NMDA encephalitis is a particular concern. Recent research has shown a minority of first episode psychosis patients have NMDA receptor antibodies (4%) but without either CSF antibodies or distinguishing clinical features.35 Thus, we do not recommend routine screening for NMDA receptor antibodies in a first episode of psychosis. This is in keeping with available evidence on the autoimmune encephalitides in general, showing that their specific clinical hallmarks of seizure, movement disorders, cognition and neuropsychiatric symptoms mark them out as a distinct group: they rarely present with acute confusion alone.
Conclusions
Delirium is a clinical diagnosis that can be made positively in most cases. The clinical assessment is paramount in diagnosing acutely confused patients. This can give a positive diagnosis of delirium or widen the net to explore alternative causes of confusion.
Supportive non-pharmacological management is well established in managing delirium or anyone with acute confusion.
There are opportunities to improve delirium prevention in those at risk, to limit its clinical impact and to communicate better its significance to patients and their families. There is hope in the future for disease-modifying treatments for dementia, increasing the importance of accurate identification of patients with delirium.
Key points
There are currently no tests to diagnose delirium positively, though clinical markers of brain frailty and physiological insult may help to identify those patients most at risk.
Delirium has its own cardinal features of fluctuations, prominent inattentiveness, and temporal association with a provoking trigger: it is not a diagnosis of exclusion.
An acute confusional state without these features of delirium should raise suspicion for an alternative or additional cause; we propose a hierarchy of investigations tailored to the individual.
Collateral history, systems review, systemic as well as neurological examination are the most informative aspects of clinical assessment of delirium.
Further reading
NICE guidelines. Delirium: prevention, diagnosis and management. Clinical guideline [CG103] 2010, last edited 2019. Accessed at https://www.nice.org.uk/guidance/conditions-and-diseases/mental-health-and-behavioural-conditions/delirium/products?GuidanceProgramme=guidelines
Carson A, Ryan T. Managing acute behavioural disturbance in a neurology ward. Pract Neurol 2010;10:67-81.
Data availability statement
No data are available.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Correction notice This article has been corrected since it was first published. A sentence in the 'Pathophysiology of delirium' section has been edited for readability.
Contributors SKA and EN contributed equally to the writing and revision of the paper.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests SKA is a co-applicant and receives royalties on patent application WO/2010/046716 ‘Neurological Autoimmune Disorders’, licensed commercially for the development of assays for LGI1 and other VGKC-complex antibodies. EN has no competing interests to declare.
Provenance and peer review Commissioned; externally peer reviewed by Martin Sadler, Plymouth, UK, Lucy Pollock, Taunton, UK and Tom Hughes, Cardiff, UK.
Other content recommended for you
- Delirium and dementia with Lewy bodies: distinct diagnoses or part of the same spectrum?
- Infectious encephalitis: mimics and chameleons
- Neurological complications of renal dialysis and transplantation
- Delirium and incident dementia in hospital patients in New South Wales, Australia: retrospective cohort study
- Delirium and the risk of developing dementia: a cohort study of 12 949 patients
- ASSESSING CONFUSED PATIENTS
- Delirium in older people
- Delirium in the elderly: a clinical review
- An unusual cause of hydrocephalus
- Cortical abnormalities on MRI: what a neurologist should know