




Abstract
The hereditary spastic paraplegias are an expanding and heterogeneous group of disorders characterized by spasticity in the lower limbs. Plasma biomarkers are needed to guide the genetic testing of spastic paraplegia. Spastic paraplegia type 5 (SPG5) is an autosomal recessive spastic paraplegia due to mutations in CYP7B1, which encodes a cytochrome P450 7α-hydroxylase implicated in cholesterol and bile acids metabolism. We developed a method based on ultra-performance liquid chromatography electrospray tandem mass spectrometry to validate two plasma 25-hydroxycholesterol (25-OHC) and 27-hydroxycholesterol (27-OHC) as diagnostic biomarkers in a cohort of 21 patients with SPG5. For 14 patients, SPG5 was initially suspected on the basis of genetic analysis, and then confirmed by increased plasma 25-OHC, 27-OHC and their ratio to total cholesterol. For seven patients, the diagnosis was initially based on elevated plasma oxysterol levels and confirmed by the identification of two causal CYP7B1 mutations. The receiver operating characteristic curves analysis showed that 25-OHC, 27-OHC and their ratio to total cholesterol discriminated between SPG5 patients and healthy controls with 100% sensitivity and specificity. Taking advantage of the robustness of these plasma oxysterols, we then conducted a phase II therapeutic trial in 12 patients and tested whether candidate molecules (atorvastatin, chenodeoxycholic acid and resveratrol) can lower plasma oxysterols and improve bile acids profile. The trial consisted of a three-period, three-treatment crossover study and the six different sequences of three treatments were randomized. Using a linear mixed effect regression model with a random intercept, we observed that atorvastatin decreased moderately plasma 27-OHC (∼30%, P < 0.001) but did not change 27-OHC to total cholesterol ratio or 25-OHC levels. We also found an abnormal bile acids profile in SPG5 patients, with significantly decreased total serum bile acids associated with a relative decrease of ursodeoxycholic and lithocholic acids compared to deoxycholic acid. Treatment with chenodeoxycholic acid restored bile acids profile in SPG5 patients. Therefore, the combination of atorvastatin and chenodeoxycholic acid may be worth considering for the treatment of SPG5 patients but the neurological benefit of these metabolic interventions remains to be evaluated in phase III therapeutic trials using clinical, imaging and/or electrophysiological outcome measures with sufficient effect sizes. Overall, our study indicates that plasma 25-OHC and 27-OHC are robust diagnostic biomarkers of SPG5 and shall be used as first-line investigations in any patient with unexplained spastic paraplegia.
Introduction
Spastic paraplegia type 5 (SPG5) (MIM #270800) is an autosomal recessive inherited spastic paraplegia (HSP) due to mutations in CYP7B1 (MIM *603711) (Tsaousidou et al., 2008; Goizet et al., 2009). The protein product of CYP7B1 is a cytochrome P450 7α-hydroxylase implicated in cholesterol metabolism. It is responsible for the 7α-hydroxylation of 27-hydroxycholesterol (27-OHC) and 25-hydroxycholesterol (25-OHC) to produce bile acid intermediates, and dehydroepiandrosterone (DHEA) to produce 7-hydroxyDHEA (Fig. 1) (Russell, 2003). Thus, SPG5 is part of the increasing group of neurodegenerative diseases due to alteration in lipid homeostasis (Lamari et al., 2013, 2015). Mutations in CYP7B1 also cause a recessive congenital bile acid synthesis defect (MIM #613812) (Setchell et al., 1998). Other diseases affecting cholesterol metabolism and/or its by-products, such as cerebrotendinous xanthomatosis and Niemann-Pick type C, may affect mainly the liver during infancy and then predominantly the CNS, depending on the relative contribution of the metabolic pathways according to age. Furthermore, oxysterols, i.e. oxygenated derivatives of cholesterol or its sterol precursors, are gaining increasing attention in the field of neuroscience (Griffiths et al., 2016). The most abundant oxysterol in brain is 24S-hydroxycholesterol (24S-OHC), which is responsible for the removal of about two-thirds of cholesterol from the brain. Conversely, the flux of 27-OHC and its metabolites to the liver can be regarded as an alternative mechanism to the classical high density lipoprotein-dependent reversed cholesterol transport (Björkhem et al., 2010).
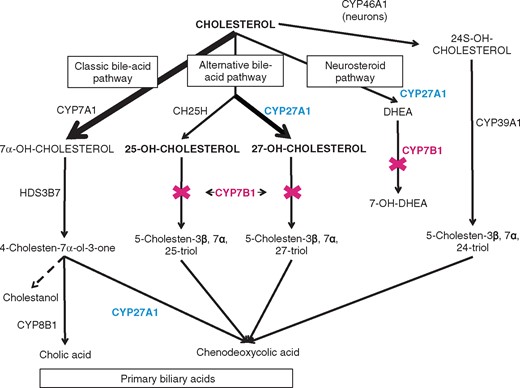
Primary bile acids simplified pathway. CYP7A1 = cholesterol 7α-hydroxylase; CYP46A1 = cholesterol 24-hydroxylase; CH25H = cholesterol 25-hydroxylase; in blue CYP27A1 = sterol 27-hydroxylase (altered in cerebrotendinous xanthomatosis); CYP7B1 = oxysterol 7α-hydroxylase (altered in SPG5); HDS3B7 = 3β-hydroxy-delta5-C27-steroid oxidoreductase; CYP8B1 = sterol 12α-hydroxylase; CYP39A1 = oxysterol7α-hydroxylase acting preferentially on 24S-OHC; DHEA = dehydroepiandrosterone.
Plasma biomarkers greatly facilitate the rapid and large scale screening of patients, especially neurological diseases with limited access to the brain itself (Gras et al., 2017). They can also be instrumental for therapeutic trials in rare diseases, which are hampered by limited number of patients, heterogeneous disease manifestations and clinical scores with small effect sizes. Several complex lipids have been validated for the diagnosis of treatable neurometabolic diseases such as plasma very long chain fatty acids for adrenoleukodystrophy, cholestanol for cerebrotendinous xanthomatosis and cholestane-3β,5α,6β-triol, 7-ketocholesterol and lyso-sphyngomyelins for Niemann-Pick type C. In cerebrotendinous xanthomatosis, the normalization of plasma cholestanol upon treatment with chenodeoxycholic acid (CDCA) is clearly associated with improved neurological functions (Nie et al., 2014). In SPG5, 25-OHC and 27-OHC have been reported to be increased in the plasma and CSF (27-OHC only) from a small series of four patients (Schüle et al., 2010). The profile of bile acids was also abnormal in the serum of these SPG5 patients (Schüle et al., 2010).
In this study, we first wished to validate plasma 25-OHC, 27-OHC and their ratio to total cholesterol as diagnostic biomarkers for SPG5 in a large series of 21 patients with CYP7B1 mutations. Taking advantage of the robustness of these biomarkers, we then conducted a phase II therapeutic trial in 12 of these patients and tested whether candidate molecules can lower plasma oxysterols and improve serum bile acids profile. We tested sequentially, but in random order: (i) atorvastatin, a well-known cholesterol-lowering drug that can decrease plasma 27-OHC in patients with moderately elevated plasma cholesterol levels (Thelen et al., 2006) and which was used in a few case reports of SPG5 patients (Mignarri et al., 2015); (ii) CDCA, which was successfully used to treat a patient with CYP7B1 mutations and severe liver disease (Dai et al., 2014); and (iii) resveratrol, which may lower cholesterol levels and exert neuroprotective properties (Liu et al., 2011; Shao et al., 2016).
Material and methods
All 21 patients presented with spastic paraplegia and were addressed to rare diseases reference centres for diagnostic and therapeutic assessment. They were over 18 years and signed a written informed consent before participating in the studies. The biomarker study was approved by the local ethical committee (RBM 01-29 SPATAX). Initially, patients benefitted either from genetic analyses (Sanger analysis or next generation sequencing panel dedicated to hereditary spastic paraplegia genes) or from biological screening (plasma 25-OHC and 27-OHC). Twelve patients from the cohort were then enrolled in a national (NCT02314208) clinical protocol promoted by INSERM and approved by the local ethical committee.
Genetic analyses
In 16 patients, the coding sequence and exon–intron boundaries of the six exons of CYP7B1 (Genbank accession number NM_004820.4) were amplified by polymerase chain reaction and analysed using direct sequencing as previously described (Goizet et al., 2009). Five patients were explored with a custom panel of 70 hereditary spastic paraplegia genes (Morais et al., 2017) (Supplementary Table 1; Roche Nimblegen) and sequenced on MiSeq machine (Illumina). Bioinformatic analyses were performed using a pipeline developed by GenoDiag society. All mutations were confirmed by Sanger sequencing. In silico analyses of missense variants were performed using four prediction models: Align GVGD (http://agvgd.hci.utah.edu/), PolyPhen-2 [Human Diversity (HumDiv) and Human Variation (HumVar) prediction models, http://genetics.bwh.harvard.edu/pph2/], Mutation Taster (http://www.mutationtaster.org/) and SIFT prediction test (http://sift.jcvi.org/). The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and the genome Aggregation Database (http://gnomad.broadinstitute.org/) were examined for single nucleotide polymorphisms (SNPs). Alamut Visual software (Interactive Biosoftware, Rouen, France) was used to perform orthologue alignments.
Biochemical analyses for plasma oxysterols and serum bile acids
Total blood was collected from fasted subjects (12 h). After centrifugation, plasma EDTA and serum were stored at −80°C and analysed within the same batch to avoid any analytical variability in the quantification of oxysterols and bile acids.
An ultra performance liquid chromatography-tandem mass spectrometer (UPLC-MS/MS) with isotopic dilution method was developed to measure plasma 25-OHC and 27-OHC. We also measured plasma 24S-OHC as decreased plasma levels have been reported in other neurodegenerative diseases (Leoni and Caccia, 2011, 2015). Preparation of EDTA plasma samples was performed according to a previously published method with slight modifications (Honda et al., 2008). Twenty microlitres of a mixture of d6-25-hydroxycholesterol, d6-27-hydroxycholesterol and d7-24S-hydroxycholesterol in methanol as internal standards and 5 µl of butylated hydroxytoluene were added to 100 µl plasma. Hydrolysis was carried out in ethanolic KOH and free oxysterols were extracted with 2 ml n-hexane and evaporated to dryness under nitrogen. The residue was derivatized to picolinyl esters by adding 200 µl of derivatizing solution containing anhydrine 2-methyl-6-nitrobenzoique, 4-dimethyl aminopyridine and picolinic acid. The mixture was incubated at 80°C for 60 min. Derivatized oxysterols were extracted with 1 ml of n-hexane and evaporated to dryness. The residue was redissolved in 100 µl methanol and an aliquot of 10 µl was injected into the UPLC-MS/MS system. The UPLC-MS/MS system consisted of a triple quadripole mass spectrometer (TQD-Waters) equipped with an electrospray ionization (ESI) probe and coupled to a UPLC Acquity system (Waters). Chromatographic separation was performed on a C-18 RP-BEH column (50 × 2.1 mm × 1.7 µl, waters) at 30°C. Eluent flow rate was set to 0.5 ml/min, the gradient of the mobile phase started with 70% of eluent A (acetonitrile/methanol/ 50:50 with 0.1% acetic acid) and 30% of eluent B (water), linear increasing to 88% of eluent A at the end of the run after 8 min. Total cholesterol was also measured to determine 25-OHC/total cholesterol and 27-OHC/total cholesterol ratios. ESI was performed in positive mode and MS/MS detection was operated in multiple-reaction monitoring mode.
Bile acids were extracted from serum by a solid-phase extraction and measured using HPLC-MS/MS. A first separation of bile acids and 7α-C4 based on polarity was carried out with an analytical column [Pinnacle II C18; Restek; 250 mm × 3.2 mm (LxID), 5 μm silica particles; Restek] fitted on a HPLC binary pump (Agilent 1100; Agilent Technologies). The mobile phase was composed of a mixture of ammonium acetate 15 mM, pH 5.3 (A) and methanol (B). HPLC was coupled with the turbo ion-spray source of the tandem MS (QTRAP 2000; Applied Biosystems/MDS SCIEX). ESI was performed in negative mode, with nitrogen as the nebulizer gas. Evaporation temperature was set at 400°C. The ion-spray, declustering and entrance potentials were set at −4500, −60, and −10 V, respectively. Collision-induced dissociation was achieved in a Q2 collision cell under various voltage potentials, depending on bile acids conjugation, and MS/MS detection was operated with unit/unit resolution in multiple-reaction-monitoring mode. Dwell time of the ion trap was set at 70 ms for each transition. Data were acquired with Analyst 1.4.2 software (AB SCIEX). The following metabolites were retained for subsequent analyses: total bile acids, CDCA, cholic acid, ursodeoxycholic acid (UDCA), lithocholic acid (LCA) and deoxycholic acid.
Phase II therapeutic trial
Exclusion criteria for the study were known hypersensitivity to atorvastatin, CDCA, resveratrol or to any of their by-products, cholesterol lowering medications other than the study treatments, elevated transaminases above three times the upper range, progressive biliary pathology, chronic diarrhoea and significant comorbid neurological disorder. Twelve patients carrying two mutations in the CYP7B1 gene were enrolled and displaying increased plasma 27-OHC and 25-OHC levels. The following doses were used for each molecule: (i) atorvastatin was administered at the standard cholesterol-lowering dose of 20 mg twice per day (Thelen et al., 2006); (ii) CDCA was administered at the dose of 500 mg twice per day, which is slightly higher than in cerebrotendinous xanthomatosis (Nie et al., 2014); and (iii) resveratrol was administered at a dose of 40 mg twice per day, based on the bioavailability reported for the specific formula that we used in this trial (Amiot et al., 2013).
The trial consisted of a three-period, three-treatment crossover study. The six different sequences of three treatments (atorvastatin, CDCA and resveratrol) were randomized: each patient received the three treatments during three periods of 2 months in random order, separated by a 4-month washout (Supplementary Fig. 1). Each sequence of treatments was administered to two patients, for a total sample size of 12 subjects. Standard lab analyses (blood electrolytes, creatinine, liver enzymes, creatine kinase, total cholesterol), as well as plasma oxysterols and serum bile acids as described above, were measured at the beginning and the end of each treatment phase as well as the washout final visit. At each visit (seven visits), we also recorded patient’s vital signs, body weight, adverse events and treatment compliance. The primary outcome measure consisted of plasma 27-OHC levels. Secondary outcome measures were: plasma 25-OHC levels, plasma oxysterols to total cholesterol ratio, serum bile acids profile, and the safety profile of the three study drugs (atorvastatin, CDCAs and resveratrol) in patients with SPG5.
Statistics
Diagnostic performance of plasma oxysterols for the diagnostic of SPG5
We performed Welch’s t-test, which accounts for unequal sample sizes and variance with Bonferroni correction. Boxplots were generated to compare the distribution of plasma oxysterols between SPG5 and controls. The performance of plasma oxysterols for the diagnostic of SPG5 was evaluated using a receiver operating characteristic (ROC) curve. Areas under the ROC curve were computed for each plasma oxysterols.
Phase II therapeutic trial
Each metabolite (oxysterols and bile acids) was evaluated under treatment with the following method. Briefly, the treatment effect between the beginning and the end of a period was analysed for each parameter using a linear mixed effect regression model with a random intercept. Treatment effect estimation was adjusted for the sequence, the period and the carry-over effect. All tests used the F statistic, and were two-tailed with a 5% significant level. All analyses were performed with R version 3.4.0 software.
Results
Characterization of the SPG5 cohort
We collected data from 21 patients with SPG5 from 18 families. Genetic data are reported in Tables 1 and 2. Five patients (24%) were born from family-related parents and 10 patients (48%) had a family history positive for spastic paraplegia. Seven patients (33%) harboured homozygous mutations. For 14 patients, the diagnosis was initially based on the identification of CYP7B1 mutations, and subsequent analysis showed that they all exhibited increased plasma levels of 25-OHC and 27-OHC. For seven patients, the diagnosis was initially based on elevated plasma oxysterols, and then confirmed by the identification of two mutations in the CYP7B1 gene.
Biochemical and genetic features of SPG5 patients (Patients 1–10)
| ID | SAL-930-001a | SAL-399-975a | GRE-506-010a | GRE-506-011a | SAL-1311-001a | SAL-1134-004a | SAL-1465-001a | NIM-001 | MON-001 | SAL-399-229a |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 12/M | 35/M | 11/M | 10/M | 20/F | 1/F | 11/M | 20/M | 40/F | 24/F |
| Origin | Algeria | France | France | France | France | France | France | France | France | Portugal |
| Consanguinity/ family history | Yes/no | No/no | No/yes | No/yes | No/no | No/no | No/no | No/yes | No/no | Yes/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes | Yes; urinary signs | Yes | Yes; saccadic pursuit; macular dystrophy; urinary signs |
| DD (years) | 14 | 30 | 28 | 26 | 15 | 28 | 35 | 35 | >10 | 39 |
| Disability scorec (/7) | NA | 6 | 3 | 3 | 2 | 5 | NA | 3 | 5 | 6 |
| 25-OHC, nmol/l | 382 | 367 | 188 | 97 | 121 | 713 | 258 | 269 | 405 | 322 |
| 27-OHC, nmol/l | 1443 | 2683 | 1331 | 1416 | 850 | 1731 | 2029 | 1736 | 1434 | 1606 |
| 24S-OHC nmol/l | 77 | 86 | 121 | 76 | 31 | 139 | 70 | 94 | 124 | 92 |
| TChol, mmol/l | 4.29 | 7.37 | 4.5 | 4.43 | 4.43 | 6.7 | 5.69 | 6 | 6 | 4.41 |
| 25-OHC/TChol | 89.0 | 49.8 | 16.8 | 21.9 | 27.3 | 106.42 | 45.3 | 48.9 | 69.8 | 73.0 |
| 27-OHC/TChol | 336.4 | 364.0 | 119 | 319.6 | 191.9 | 258.36 | 356.6 | 315.6 | 247 | 364.2 |
| 24S-OHC/TChol | 17.9 | 11.7 | 26.9 | 17.2 | 7 | 20.7 | 12.3 | 17.1 | 21.4 | 20.9 |
| Mutation cDNA | c.1162C> T (homoz) | c.1250G>A | c.889A>G | c.889A>G | c.308_309insA | c.392dup | c.1061G>A | c.440G>A | c.1163G>A | c.524G>A (homoz) |
| c.1456C>T | c.1061G>A | c.1061G>A | c.1456C>T | c.825T>A | c.1456C>T | c.1193C>T | c.1355G>A | |||
| c.889A>G | ||||||||||
| Protein (predicted) | p.Arg388* | p.Arg417His | p.Thr297Ala | p.Thr297Ala | p.Asn105Phefs*3 | p.Asn131Lysfs*3 | p.Ser354Asn | p.Gly147Asp | p.Arg388Gln | p.Trp175* |
| p.Arg486Cys | p.Ser354Asn | p.Ser354Asn | p.Arg486Cys | p.Tyr275* | p.Arg486Cys | p.Pro398Leu | p.Arg452Gln | |||
| p.Thr297Ala | ||||||||||
| Novel/known mutation | Knownd,e | Knownd,f,g,h | Knownf,h,j | Knownf,h,j | Knownk | Novel | Novel | Knowni | Novel | Novel |
| Knownf,h,i | Novel | Novel | Knownf,h,i | Knownf | Knownf,h,i | Novel | ||||
| Knownf,h,j | ||||||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | NA | Frame shift | Conserved up to 12 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 7 species, deleterious (PP only), frequency 2/252 394 (gnomAD) | Deleterious (SF, PP), deleterious (SF, PP) | Nonsense mutation, absent from gnomAD |
| ID | SAL-930-001a | SAL-399-975a | GRE-506-010a | GRE-506-011a | SAL-1311-001a | SAL-1134-004a | SAL-1465-001a | NIM-001 | MON-001 | SAL-399-229a |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 12/M | 35/M | 11/M | 10/M | 20/F | 1/F | 11/M | 20/M | 40/F | 24/F |
| Origin | Algeria | France | France | France | France | France | France | France | France | Portugal |
| Consanguinity/ family history | Yes/no | No/no | No/yes | No/yes | No/no | No/no | No/no | No/yes | No/no | Yes/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes | Yes; urinary signs | Yes | Yes; saccadic pursuit; macular dystrophy; urinary signs |
| DD (years) | 14 | 30 | 28 | 26 | 15 | 28 | 35 | 35 | >10 | 39 |
| Disability scorec (/7) | NA | 6 | 3 | 3 | 2 | 5 | NA | 3 | 5 | 6 |
| 25-OHC, nmol/l | 382 | 367 | 188 | 97 | 121 | 713 | 258 | 269 | 405 | 322 |
| 27-OHC, nmol/l | 1443 | 2683 | 1331 | 1416 | 850 | 1731 | 2029 | 1736 | 1434 | 1606 |
| 24S-OHC nmol/l | 77 | 86 | 121 | 76 | 31 | 139 | 70 | 94 | 124 | 92 |
| TChol, mmol/l | 4.29 | 7.37 | 4.5 | 4.43 | 4.43 | 6.7 | 5.69 | 6 | 6 | 4.41 |
| 25-OHC/TChol | 89.0 | 49.8 | 16.8 | 21.9 | 27.3 | 106.42 | 45.3 | 48.9 | 69.8 | 73.0 |
| 27-OHC/TChol | 336.4 | 364.0 | 119 | 319.6 | 191.9 | 258.36 | 356.6 | 315.6 | 247 | 364.2 |
| 24S-OHC/TChol | 17.9 | 11.7 | 26.9 | 17.2 | 7 | 20.7 | 12.3 | 17.1 | 21.4 | 20.9 |
| Mutation cDNA | c.1162C> T (homoz) | c.1250G>A | c.889A>G | c.889A>G | c.308_309insA | c.392dup | c.1061G>A | c.440G>A | c.1163G>A | c.524G>A (homoz) |
| c.1456C>T | c.1061G>A | c.1061G>A | c.1456C>T | c.825T>A | c.1456C>T | c.1193C>T | c.1355G>A | |||
| c.889A>G | ||||||||||
| Protein (predicted) | p.Arg388* | p.Arg417His | p.Thr297Ala | p.Thr297Ala | p.Asn105Phefs*3 | p.Asn131Lysfs*3 | p.Ser354Asn | p.Gly147Asp | p.Arg388Gln | p.Trp175* |
| p.Arg486Cys | p.Ser354Asn | p.Ser354Asn | p.Arg486Cys | p.Tyr275* | p.Arg486Cys | p.Pro398Leu | p.Arg452Gln | |||
| p.Thr297Ala | ||||||||||
| Novel/known mutation | Knownd,e | Knownd,f,g,h | Knownf,h,j | Knownf,h,j | Knownk | Novel | Novel | Knowni | Novel | Novel |
| Knownf,h,i | Novel | Novel | Knownf,h,i | Knownf | Knownf,h,i | Novel | ||||
| Knownf,h,j | ||||||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | NA | Frame shift | Conserved up to 12 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 7 species, deleterious (PP only), frequency 2/252 394 (gnomAD) | Deleterious (SF, PP), deleterious (SF, PP) | Nonsense mutation, absent from gnomAD |
aPatients who were enrolled in the therapeutic trial.
bLower limb spasticity and four limbs deep sensation alteration.
cDisability score (0 = no functional handicap, 1 = no functional handicap but signs at examination; 2 = mild, able to run, walking unlimited; 3 = moderate, unable to run, limited walking without aid; 4 = severe, walking with one stick; 5 = walking with two sticks; 6 = unable to walk, requiring wheelchair; 7 = confined to bed).
dTsaousidou et al., 2008; eSetchell et al., 1998; fGoizet et al., 2009; gVanotti et al., 2014; hSiam et al., 2012; iRoos et al., 2014; jDi Fabio et al., 2014; kSchüle et al., 2009.
DD = disease duration; homoz = homozygous; MT = Mutation Taster; PP = PolyPhen-2; TChol = total cholesterol; SF = SIFT.
Biochemical and genetic features of SPG5 patients (Patients 1–10)
| ID | SAL-930-001a | SAL-399-975a | GRE-506-010a | GRE-506-011a | SAL-1311-001a | SAL-1134-004a | SAL-1465-001a | NIM-001 | MON-001 | SAL-399-229a |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 12/M | 35/M | 11/M | 10/M | 20/F | 1/F | 11/M | 20/M | 40/F | 24/F |
| Origin | Algeria | France | France | France | France | France | France | France | France | Portugal |
| Consanguinity/ family history | Yes/no | No/no | No/yes | No/yes | No/no | No/no | No/no | No/yes | No/no | Yes/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes | Yes; urinary signs | Yes | Yes; saccadic pursuit; macular dystrophy; urinary signs |
| DD (years) | 14 | 30 | 28 | 26 | 15 | 28 | 35 | 35 | >10 | 39 |
| Disability scorec (/7) | NA | 6 | 3 | 3 | 2 | 5 | NA | 3 | 5 | 6 |
| 25-OHC, nmol/l | 382 | 367 | 188 | 97 | 121 | 713 | 258 | 269 | 405 | 322 |
| 27-OHC, nmol/l | 1443 | 2683 | 1331 | 1416 | 850 | 1731 | 2029 | 1736 | 1434 | 1606 |
| 24S-OHC nmol/l | 77 | 86 | 121 | 76 | 31 | 139 | 70 | 94 | 124 | 92 |
| TChol, mmol/l | 4.29 | 7.37 | 4.5 | 4.43 | 4.43 | 6.7 | 5.69 | 6 | 6 | 4.41 |
| 25-OHC/TChol | 89.0 | 49.8 | 16.8 | 21.9 | 27.3 | 106.42 | 45.3 | 48.9 | 69.8 | 73.0 |
| 27-OHC/TChol | 336.4 | 364.0 | 119 | 319.6 | 191.9 | 258.36 | 356.6 | 315.6 | 247 | 364.2 |
| 24S-OHC/TChol | 17.9 | 11.7 | 26.9 | 17.2 | 7 | 20.7 | 12.3 | 17.1 | 21.4 | 20.9 |
| Mutation cDNA | c.1162C> T (homoz) | c.1250G>A | c.889A>G | c.889A>G | c.308_309insA | c.392dup | c.1061G>A | c.440G>A | c.1163G>A | c.524G>A (homoz) |
| c.1456C>T | c.1061G>A | c.1061G>A | c.1456C>T | c.825T>A | c.1456C>T | c.1193C>T | c.1355G>A | |||
| c.889A>G | ||||||||||
| Protein (predicted) | p.Arg388* | p.Arg417His | p.Thr297Ala | p.Thr297Ala | p.Asn105Phefs*3 | p.Asn131Lysfs*3 | p.Ser354Asn | p.Gly147Asp | p.Arg388Gln | p.Trp175* |
| p.Arg486Cys | p.Ser354Asn | p.Ser354Asn | p.Arg486Cys | p.Tyr275* | p.Arg486Cys | p.Pro398Leu | p.Arg452Gln | |||
| p.Thr297Ala | ||||||||||
| Novel/known mutation | Knownd,e | Knownd,f,g,h | Knownf,h,j | Knownf,h,j | Knownk | Novel | Novel | Knowni | Novel | Novel |
| Knownf,h,i | Novel | Novel | Knownf,h,i | Knownf | Knownf,h,i | Novel | ||||
| Knownf,h,j | ||||||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | NA | Frame shift | Conserved up to 12 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 7 species, deleterious (PP only), frequency 2/252 394 (gnomAD) | Deleterious (SF, PP), deleterious (SF, PP) | Nonsense mutation, absent from gnomAD |
| ID | SAL-930-001a | SAL-399-975a | GRE-506-010a | GRE-506-011a | SAL-1311-001a | SAL-1134-004a | SAL-1465-001a | NIM-001 | MON-001 | SAL-399-229a |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 12/M | 35/M | 11/M | 10/M | 20/F | 1/F | 11/M | 20/M | 40/F | 24/F |
| Origin | Algeria | France | France | France | France | France | France | France | France | Portugal |
| Consanguinity/ family history | Yes/no | No/no | No/yes | No/yes | No/no | No/no | No/no | No/yes | No/no | Yes/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes | Yes; urinary signs | Yes | Yes; saccadic pursuit; macular dystrophy; urinary signs |
| DD (years) | 14 | 30 | 28 | 26 | 15 | 28 | 35 | 35 | >10 | 39 |
| Disability scorec (/7) | NA | 6 | 3 | 3 | 2 | 5 | NA | 3 | 5 | 6 |
| 25-OHC, nmol/l | 382 | 367 | 188 | 97 | 121 | 713 | 258 | 269 | 405 | 322 |
| 27-OHC, nmol/l | 1443 | 2683 | 1331 | 1416 | 850 | 1731 | 2029 | 1736 | 1434 | 1606 |
| 24S-OHC nmol/l | 77 | 86 | 121 | 76 | 31 | 139 | 70 | 94 | 124 | 92 |
| TChol, mmol/l | 4.29 | 7.37 | 4.5 | 4.43 | 4.43 | 6.7 | 5.69 | 6 | 6 | 4.41 |
| 25-OHC/TChol | 89.0 | 49.8 | 16.8 | 21.9 | 27.3 | 106.42 | 45.3 | 48.9 | 69.8 | 73.0 |
| 27-OHC/TChol | 336.4 | 364.0 | 119 | 319.6 | 191.9 | 258.36 | 356.6 | 315.6 | 247 | 364.2 |
| 24S-OHC/TChol | 17.9 | 11.7 | 26.9 | 17.2 | 7 | 20.7 | 12.3 | 17.1 | 21.4 | 20.9 |
| Mutation cDNA | c.1162C> T (homoz) | c.1250G>A | c.889A>G | c.889A>G | c.308_309insA | c.392dup | c.1061G>A | c.440G>A | c.1163G>A | c.524G>A (homoz) |
| c.1456C>T | c.1061G>A | c.1061G>A | c.1456C>T | c.825T>A | c.1456C>T | c.1193C>T | c.1355G>A | |||
| c.889A>G | ||||||||||
| Protein (predicted) | p.Arg388* | p.Arg417His | p.Thr297Ala | p.Thr297Ala | p.Asn105Phefs*3 | p.Asn131Lysfs*3 | p.Ser354Asn | p.Gly147Asp | p.Arg388Gln | p.Trp175* |
| p.Arg486Cys | p.Ser354Asn | p.Ser354Asn | p.Arg486Cys | p.Tyr275* | p.Arg486Cys | p.Pro398Leu | p.Arg452Gln | |||
| p.Thr297Ala | ||||||||||
| Novel/known mutation | Knownd,e | Knownd,f,g,h | Knownf,h,j | Knownf,h,j | Knownk | Novel | Novel | Knowni | Novel | Novel |
| Knownf,h,i | Novel | Novel | Knownf,h,i | Knownf | Knownf,h,i | Novel | ||||
| Knownf,h,j | ||||||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | NA | Frame shift | Conserved up to 12 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) | Conserved up to 7 species, deleterious (PP only), frequency 2/252 394 (gnomAD) | Deleterious (SF, PP), deleterious (SF, PP) | Nonsense mutation, absent from gnomAD |
aPatients who were enrolled in the therapeutic trial.
bLower limb spasticity and four limbs deep sensation alteration.
cDisability score (0 = no functional handicap, 1 = no functional handicap but signs at examination; 2 = mild, able to run, walking unlimited; 3 = moderate, unable to run, limited walking without aid; 4 = severe, walking with one stick; 5 = walking with two sticks; 6 = unable to walk, requiring wheelchair; 7 = confined to bed).
dTsaousidou et al., 2008; eSetchell et al., 1998; fGoizet et al., 2009; gVanotti et al., 2014; hSiam et al., 2012; iRoos et al., 2014; jDi Fabio et al., 2014; kSchüle et al., 2009.
DD = disease duration; homoz = homozygous; MT = Mutation Taster; PP = PolyPhen-2; TChol = total cholesterol; SF = SIFT.
Biochemical and genetic features of SPG5 patients (Patients 11 to 21)
| ID | SAL-279-014a | SAL-B-563-010a | SAL-D-563-010 | SAL-004-006a | SAL-004-008 | SAL-004-015 | SAL-1450-001a | SAL-1491-001 | SAL-899-027 | BEL-001 | LYO-001 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 16/M | 10/F | 44/F | 20/M | 27/M | 7/F | 40/F | 10/M | 37/F | 16/F | NA/F |
| Origin | France | France | Portugal | France | France | France | France | Algeria | France | Belgium | France |
| Consanguinity/ family history | No/no | No/no | Yes/yes | No/yes | No/yes | No/yes | No/yes | Yes/yes | Yes/yes | No/no | No/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit; optic atrophy; urinary signs | Yes; urinary signs | Yes; saccadic pursuit, nyst; urinary signs; cerebellar ataxia, myocl | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; demyelin PNP, urinary signs | Yes | Yes; urinary signs | Yes |
| DD (years) | 42 | 50 | 25 | 34 | NA | 27 | 17 | 37 | 34 | 18 | >20 |
| Disability scorec (/7) | 5 | 4 | 4 | 6 | 6 | 6 | 3 | 3 | 6 | 5 | NA |
| 25-OHC, nmol/l | 189 | 106 | 134 | 160 | 86 | 174 | 114 | 386 | 281 | 91 | 1149 |
| 27-OHC, nmol/l | 1299 | 764 | 812 | 1362 | 2269 | 1017 | 1530 | 1938 | 1804 | 2120 | 2017 |
| 24S-OHC nmol/l | 69 | 82 | 67 | 55 | NA | 95 | 139 | 63 | 142 | 85 | 61 |
| Tchol, mmol/l | 5.0 | 4.6 | 4.0 | 3.4 | 3 | 5.1 | 5.7 | 5.4 | 6.6 | 4.1 | 6.4 |
| 25-OHC/ TChol | 37.6 | 22.9 | 33.3 | 47.3 | 31.2 | 33.9 | 20.0 | 72.0 | 42.6 | 22.4 | 180.9 |
| 27-OHC/ TChol | 258.3 | 165.8 | 202.1 | 404.2 | 822.1 | 198.2 | 268.0 | 362.0 | 273.3 | 520.9 | 317.6 |
| 24S-OHC/ TChol | 13.7 | 17.8 | 16.7 | 16.3 | NA | 18.5 | 24.3 | 11.8 | 21.5 | 20.9 | 9.6 |
| Mutation cDNA | c.187C>T | c.889A>G (homoz) | c.1456C>T (homoz) | c.1250G>A | c.1250G>A | c.1250G>A | c.440G-A (homoz) | c.1162C>T (homoz) | c.1456C>T (homoz) | c.334C>T | c.260-1G>A |
| c.889A>G | c.1408T>A | c.1408T>A | c.1408T>A | c.825T>A | c.1061G>A | ||||||
| Protein (predicted) | p.Arg63* | p.Thr 297Ala | p.Arg486Cys | p.Arg 417His | p.Arg 417His | p.Arg417His | p.Gly147Asp | p.Arg 388* | p.Arg 486Cys | p.R112* | Splice site mutation |
| p.Thr297Ala | p.Phe470Ile | p.Phe470Ile | p.Phe470Ile | p.Tyr275* | p.Ser354Asn | ||||||
| Novel/known mutation | Knownd | Knownd,e,f | Knownd,e,g | Knownd,e,h,i | Knownd,e,h,i | Knownd,e,h,i | Knowng | Knownh,j | Knownd,e,g | Knownd,f,g | Novel |
| Knownd,e,f | Knownd | Knownd | Knownd | Knownd | Novel | ||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Splicing mutation, conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) |
| ID | SAL-279-014a | SAL-B-563-010a | SAL-D-563-010 | SAL-004-006a | SAL-004-008 | SAL-004-015 | SAL-1450-001a | SAL-1491-001 | SAL-899-027 | BEL-001 | LYO-001 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 16/M | 10/F | 44/F | 20/M | 27/M | 7/F | 40/F | 10/M | 37/F | 16/F | NA/F |
| Origin | France | France | Portugal | France | France | France | France | Algeria | France | Belgium | France |
| Consanguinity/ family history | No/no | No/no | Yes/yes | No/yes | No/yes | No/yes | No/yes | Yes/yes | Yes/yes | No/no | No/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit; optic atrophy; urinary signs | Yes; urinary signs | Yes; saccadic pursuit, nyst; urinary signs; cerebellar ataxia, myocl | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; demyelin PNP, urinary signs | Yes | Yes; urinary signs | Yes |
| DD (years) | 42 | 50 | 25 | 34 | NA | 27 | 17 | 37 | 34 | 18 | >20 |
| Disability scorec (/7) | 5 | 4 | 4 | 6 | 6 | 6 | 3 | 3 | 6 | 5 | NA |
| 25-OHC, nmol/l | 189 | 106 | 134 | 160 | 86 | 174 | 114 | 386 | 281 | 91 | 1149 |
| 27-OHC, nmol/l | 1299 | 764 | 812 | 1362 | 2269 | 1017 | 1530 | 1938 | 1804 | 2120 | 2017 |
| 24S-OHC nmol/l | 69 | 82 | 67 | 55 | NA | 95 | 139 | 63 | 142 | 85 | 61 |
| Tchol, mmol/l | 5.0 | 4.6 | 4.0 | 3.4 | 3 | 5.1 | 5.7 | 5.4 | 6.6 | 4.1 | 6.4 |
| 25-OHC/ TChol | 37.6 | 22.9 | 33.3 | 47.3 | 31.2 | 33.9 | 20.0 | 72.0 | 42.6 | 22.4 | 180.9 |
| 27-OHC/ TChol | 258.3 | 165.8 | 202.1 | 404.2 | 822.1 | 198.2 | 268.0 | 362.0 | 273.3 | 520.9 | 317.6 |
| 24S-OHC/ TChol | 13.7 | 17.8 | 16.7 | 16.3 | NA | 18.5 | 24.3 | 11.8 | 21.5 | 20.9 | 9.6 |
| Mutation cDNA | c.187C>T | c.889A>G (homoz) | c.1456C>T (homoz) | c.1250G>A | c.1250G>A | c.1250G>A | c.440G-A (homoz) | c.1162C>T (homoz) | c.1456C>T (homoz) | c.334C>T | c.260-1G>A |
| c.889A>G | c.1408T>A | c.1408T>A | c.1408T>A | c.825T>A | c.1061G>A | ||||||
| Protein (predicted) | p.Arg63* | p.Thr 297Ala | p.Arg486Cys | p.Arg 417His | p.Arg 417His | p.Arg417His | p.Gly147Asp | p.Arg 388* | p.Arg 486Cys | p.R112* | Splice site mutation |
| p.Thr297Ala | p.Phe470Ile | p.Phe470Ile | p.Phe470Ile | p.Tyr275* | p.Ser354Asn | ||||||
| Novel/known mutation | Knownd | Knownd,e,f | Knownd,e,g | Knownd,e,h,i | Knownd,e,h,i | Knownd,e,h,i | Knowng | Knownh,j | Knownd,e,g | Knownd,f,g | Novel |
| Knownd,e,f | Knownd | Knownd | Knownd | Knownd | Novel | ||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Splicing mutation, conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) |
aPatients who were enrolled in the therapeutic trial.
bLower limb spasticity and four limbs deep sensation alteration.
cDisability score (0 = no functional handicap, 1 = no functional handicap but signs at examination; 2 = mild, able to run, walking unlimited; 3 = moderate, unable to run, limited walking without aid; 4 = severe, walking with one stick; 5 = walking with two sticks; 6 = unable to walk, requiring wheelchair; 7 = confined to bed).
dGoizet et al., 2009; eSiam et al., 2012; fDi Fabio et al., 2014; gRoos et al., 2014; hTsaousidou et al., 2008; iVanotti et al., 2014; jSetchell et al., 1998.
DD = disease duration; demyel PNP = demyelinating polyneuropathy; homoz = homozygous; myocl = myoclonus; MT = MutationTaster; nyst = nystagmus; PP = PolyPhen-2; SF = SIFT; TChol = total cholesterol.
Biochemical and genetic features of SPG5 patients (Patients 11 to 21)
| ID | SAL-279-014a | SAL-B-563-010a | SAL-D-563-010 | SAL-004-006a | SAL-004-008 | SAL-004-015 | SAL-1450-001a | SAL-1491-001 | SAL-899-027 | BEL-001 | LYO-001 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 16/M | 10/F | 44/F | 20/M | 27/M | 7/F | 40/F | 10/M | 37/F | 16/F | NA/F |
| Origin | France | France | Portugal | France | France | France | France | Algeria | France | Belgium | France |
| Consanguinity/ family history | No/no | No/no | Yes/yes | No/yes | No/yes | No/yes | No/yes | Yes/yes | Yes/yes | No/no | No/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit; optic atrophy; urinary signs | Yes; urinary signs | Yes; saccadic pursuit, nyst; urinary signs; cerebellar ataxia, myocl | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; demyelin PNP, urinary signs | Yes | Yes; urinary signs | Yes |
| DD (years) | 42 | 50 | 25 | 34 | NA | 27 | 17 | 37 | 34 | 18 | >20 |
| Disability scorec (/7) | 5 | 4 | 4 | 6 | 6 | 6 | 3 | 3 | 6 | 5 | NA |
| 25-OHC, nmol/l | 189 | 106 | 134 | 160 | 86 | 174 | 114 | 386 | 281 | 91 | 1149 |
| 27-OHC, nmol/l | 1299 | 764 | 812 | 1362 | 2269 | 1017 | 1530 | 1938 | 1804 | 2120 | 2017 |
| 24S-OHC nmol/l | 69 | 82 | 67 | 55 | NA | 95 | 139 | 63 | 142 | 85 | 61 |
| Tchol, mmol/l | 5.0 | 4.6 | 4.0 | 3.4 | 3 | 5.1 | 5.7 | 5.4 | 6.6 | 4.1 | 6.4 |
| 25-OHC/ TChol | 37.6 | 22.9 | 33.3 | 47.3 | 31.2 | 33.9 | 20.0 | 72.0 | 42.6 | 22.4 | 180.9 |
| 27-OHC/ TChol | 258.3 | 165.8 | 202.1 | 404.2 | 822.1 | 198.2 | 268.0 | 362.0 | 273.3 | 520.9 | 317.6 |
| 24S-OHC/ TChol | 13.7 | 17.8 | 16.7 | 16.3 | NA | 18.5 | 24.3 | 11.8 | 21.5 | 20.9 | 9.6 |
| Mutation cDNA | c.187C>T | c.889A>G (homoz) | c.1456C>T (homoz) | c.1250G>A | c.1250G>A | c.1250G>A | c.440G-A (homoz) | c.1162C>T (homoz) | c.1456C>T (homoz) | c.334C>T | c.260-1G>A |
| c.889A>G | c.1408T>A | c.1408T>A | c.1408T>A | c.825T>A | c.1061G>A | ||||||
| Protein (predicted) | p.Arg63* | p.Thr 297Ala | p.Arg486Cys | p.Arg 417His | p.Arg 417His | p.Arg417His | p.Gly147Asp | p.Arg 388* | p.Arg 486Cys | p.R112* | Splice site mutation |
| p.Thr297Ala | p.Phe470Ile | p.Phe470Ile | p.Phe470Ile | p.Tyr275* | p.Ser354Asn | ||||||
| Novel/known mutation | Knownd | Knownd,e,f | Knownd,e,g | Knownd,e,h,i | Knownd,e,h,i | Knownd,e,h,i | Knowng | Knownh,j | Knownd,e,g | Knownd,f,g | Novel |
| Knownd,e,f | Knownd | Knownd | Knownd | Knownd | Novel | ||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Splicing mutation, conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) |
| ID | SAL-279-014a | SAL-B-563-010a | SAL-D-563-010 | SAL-004-006a | SAL-004-008 | SAL-004-015 | SAL-1450-001a | SAL-1491-001 | SAL-899-027 | BEL-001 | LYO-001 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset/sex | 16/M | 10/F | 44/F | 20/M | 27/M | 7/F | 40/F | 10/M | 37/F | 16/F | NA/F |
| Origin | France | France | Portugal | France | France | France | France | Algeria | France | Belgium | France |
| Consanguinity/ family history | No/no | No/no | Yes/yes | No/yes | No/yes | No/yes | No/yes | Yes/yes | Yes/yes | No/no | No/no |
| Global common phenotypeb; additional signs | Yes; saccadic pursuit | Yes; saccadic pursuit; urinary signs | Yes; saccadic pursuit; optic atrophy; urinary signs | Yes; urinary signs | Yes; saccadic pursuit, nyst; urinary signs; cerebellar ataxia, myocl | Yes; urinary signs | Yes; saccadic pursuit; urinary signs | Yes; demyelin PNP, urinary signs | Yes | Yes; urinary signs | Yes |
| DD (years) | 42 | 50 | 25 | 34 | NA | 27 | 17 | 37 | 34 | 18 | >20 |
| Disability scorec (/7) | 5 | 4 | 4 | 6 | 6 | 6 | 3 | 3 | 6 | 5 | NA |
| 25-OHC, nmol/l | 189 | 106 | 134 | 160 | 86 | 174 | 114 | 386 | 281 | 91 | 1149 |
| 27-OHC, nmol/l | 1299 | 764 | 812 | 1362 | 2269 | 1017 | 1530 | 1938 | 1804 | 2120 | 2017 |
| 24S-OHC nmol/l | 69 | 82 | 67 | 55 | NA | 95 | 139 | 63 | 142 | 85 | 61 |
| Tchol, mmol/l | 5.0 | 4.6 | 4.0 | 3.4 | 3 | 5.1 | 5.7 | 5.4 | 6.6 | 4.1 | 6.4 |
| 25-OHC/ TChol | 37.6 | 22.9 | 33.3 | 47.3 | 31.2 | 33.9 | 20.0 | 72.0 | 42.6 | 22.4 | 180.9 |
| 27-OHC/ TChol | 258.3 | 165.8 | 202.1 | 404.2 | 822.1 | 198.2 | 268.0 | 362.0 | 273.3 | 520.9 | 317.6 |
| 24S-OHC/ TChol | 13.7 | 17.8 | 16.7 | 16.3 | NA | 18.5 | 24.3 | 11.8 | 21.5 | 20.9 | 9.6 |
| Mutation cDNA | c.187C>T | c.889A>G (homoz) | c.1456C>T (homoz) | c.1250G>A | c.1250G>A | c.1250G>A | c.440G-A (homoz) | c.1162C>T (homoz) | c.1456C>T (homoz) | c.334C>T | c.260-1G>A |
| c.889A>G | c.1408T>A | c.1408T>A | c.1408T>A | c.825T>A | c.1061G>A | ||||||
| Protein (predicted) | p.Arg63* | p.Thr 297Ala | p.Arg486Cys | p.Arg 417His | p.Arg 417His | p.Arg417His | p.Gly147Asp | p.Arg 388* | p.Arg 486Cys | p.R112* | Splice site mutation |
| p.Thr297Ala | p.Phe470Ile | p.Phe470Ile | p.Phe470Ile | p.Tyr275* | p.Ser354Asn | ||||||
| Novel/known mutation | Knownd | Knownd,e,f | Knownd,e,g | Knownd,e,h,i | Knownd,e,h,i | Knownd,e,h,i | Knowng | Knownh,j | Knownd,e,g | Knownd,f,g | Novel |
| Knownd,e,f | Knownd | Knownd | Knownd | Knownd | Novel | ||||||
| Arguments for variant pathogenic mutation (if novel) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Splicing mutation, conserved up to 11 species, deleterious (MT, PP, SF), frequency 1/30 244 (gnomAD) |
aPatients who were enrolled in the therapeutic trial.
bLower limb spasticity and four limbs deep sensation alteration.
cDisability score (0 = no functional handicap, 1 = no functional handicap but signs at examination; 2 = mild, able to run, walking unlimited; 3 = moderate, unable to run, limited walking without aid; 4 = severe, walking with one stick; 5 = walking with two sticks; 6 = unable to walk, requiring wheelchair; 7 = confined to bed).
dGoizet et al., 2009; eSiam et al., 2012; fDi Fabio et al., 2014; gRoos et al., 2014; hTsaousidou et al., 2008; iVanotti et al., 2014; jSetchell et al., 1998.
DD = disease duration; demyel PNP = demyelinating polyneuropathy; homoz = homozygous; myocl = myoclonus; MT = MutationTaster; nyst = nystagmus; PP = PolyPhen-2; SF = SIFT; TChol = total cholesterol.
Clinical presentation
A summary of the clinical phenotype of each patient is presented in Tables 1 and 2, and more details are reported in Supplementary Tables 2 and 3. Age at onset ranged from 1 to 44 years with a mean of 20 ± 13 years. After a mean disease duration of 29 ± 10 years (range 14–50), all patients exhibited a moderate-to-severe spastic paraplegia. About half of them had a severe handicap, being wheelchair-bound (disability score = 6; n = 6/21, 28.5%) or requiring one or two sticks for walking (disability score = 4 or 5; n = 7/21, 33%), but none were confined to bed, even after a very long-lasting disease. One patient was still able to walk without help (disability score = 2) about 15 years after disease onset. Neither age at onset, disability score nor disease duration correlated with plasma oxysterol levels (data not shown). The most frequent presenting symptom was leg stiffness (17/19, 89%), variably associated with unsteadiness and cramps. In a minority of patients, unsteadiness was the presenting sign (2/19, 11%). All patients developed spastic paraplegia associated with severely reduced or abolished vibration sense in the lower limbs and positive Romberg signs. Vibration sense was also reduced in the upper limbs of most patients (12/16, 75%). Two-thirds of patients reported urinary urgency or incontinence. Lower limb spasticity was severe in 11/20 patients (55%). Moreover, spasticity was often present at rest (10/15, 67%), although milder, but rarely associated with severe proximal weakness (3/20, 15%). Upper limb reflexes were increased or diffused in 11/19 patients (58%). There was no sign of cerebellar dysfunction except for slight saccadic pursuit in 10/17 patients (59%). Atypical presentations included demyelinating polyneuropathy, optic atrophy, and familial macular dystrophy. These symptoms occurred in the context of multiple consanguineous unions with the possible co-occurrence of different genetic diseases.
Electrophysiological and imaging features
Electrophysiological and imaging data were analysed retrospectively. An EMG was performed in 13/20 patients and was normal in 11 (85%). Sensory evoked potentials were available only for four patients but were always altered, with central sensory abnormalities. Cerebral MRI was available for 18/21 patients. A mild but uniform hyperintensity on T2- and FLAIR-weighted images was present in the posterior periventricular white matter and centrum semiovale of 12 patients (67%) (Supplementary Fig. 2). One patient presented with both isolated and confluent hyperintensities. Seven patients (39%) displayed mild cerebellar atrophy and only two showed mild cortical atrophy. Medullary MRI was available for 12/21 patients and spinal atrophy was observed in four patients (Supplementary Fig. 2 and Supplementary Tables 2 and 3). One patient displayed a probable syringomyelia while another patient showed an extensive posterior cordonal hyperintensity, interpreted as a subacute combined spinal degeneration in the context of moderate hyperhomocysteinaemia (28.4 μmol/l; normal < 14 μmol/l) and folate deficiency (1.5 ng/ml; normal = 3.5–17.5 ng/ml).
Extra-neurological features
Following previous reports of abnormal bile acids profile in a few patients with SPG5, we compared serum bile acids from 12 SPG5 patients to 18 controls of similar age, sex, and body mass index. We observed an abnormal bile acids profile in the serum of patients compared to controls, with decreased levels of total bile acids (P = 0.008, Fig. 2A), associated with a relative decrease of secondary bile acids derived from CDCA [UDCA (P = 0.002, Fig. 2B) and LCA (P = 0.048, Fig. 2B)] while deoxycholic acid, derived from cholic acid, showed an opposite trend, although not significant (Fig. 2C).
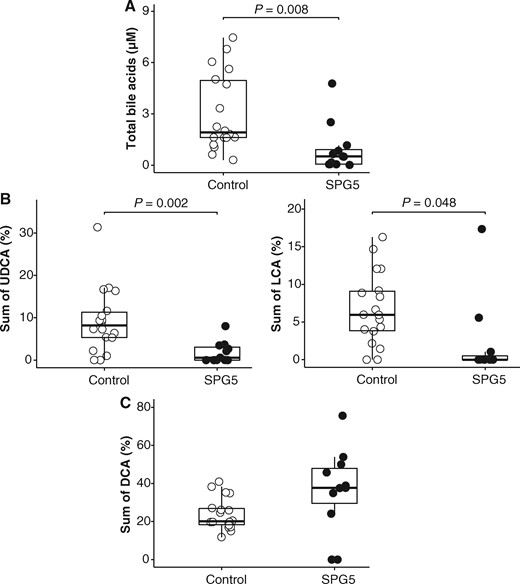
Abnormal bile acids profile in the serum of SPG5 patients. (A) Decreased levels of total bile acids in patients' serum compared with controls. Compared to controls, serum UDCA and LCA—secondary bile acids derived from CDCA—are decreased in SPG5 patients (B) while deoxycholic acid (DCA)—secondary bile acids derived from cholic acid—tend to be increased (C).
Since 27-OHC is an endogenous selective oestrogen receptor modulator, we also investigated the liver (abdominal ultrasound, hepatic fibroscan), bone (bone mineral density scan, parathyroid hormone and C-terminal telopeptide), and cardiovascular functions (cardiac transthoracic echography, supra-aortic trunk and lower limb vessel Doppler echography) of eight patients with SPG5. Liver enzymes were mildly elevated in 4/8 patients, associated with mild hepatomegaly in two patients, abnormal biliary tracts in one patient and autoimmune liver disease in another patient with abnormal fibroscan (Patient SAL-D-563-010, previously reported as Patient FSP-563-010) (Goizet et al., 2009). Atherosclerotic lesions were identified in three patients while 7/8 patients exhibited reduced bone mineral density (osteoporosis in two patients, 52 and 54 years old) with no major alterations in vitamin D, parathyroid hormone, calcium, and phosphorus concentrations.
Validation of plasma oxysterols as diagnostic markers in SPG5
Upon validation of our UPLC-MS/MS method (Supplementary material), we measured 25-OHC, 27-OHC and 24S-OHC in plasma samples from 21 patients with SPG5 (mean age 49.5 ± 14; range 24–73 years), and 37 controls of similar age (Fig. 3). In SPG5 patients, both 25-OHC (SPG5 mean: 285.3 ± 249.3 nmol/l; control mean: 15.3 ± 17.5 nmol/l; P < 0.001) and 27-OHC levels (SPG5 mean: 1580 ± 498 nmol/l; control mean: 139.3 ± 76.3 nmol/l; P < 0.001) were significantly elevated compared to control subjects (Fig. 3A). The ratios of 25-OHC and 27-OHC to total cholesterol were also higher in plasma from SPG5 patients compared to controls (P < 0.001, Fig. 3B). The ROC curves analysis showed that 25-OHC (cut-off = 66.1 nmol/l), 27-OHC (cut-off = 623 nmol/l), 25-OHC/total cholesterol (cut-off = 14.1) and 27-OHC/total cholesterol (cut-off = 95.8) discriminated SPG5 from healthy controls with 100% sensitivity and specificity (area under ROC curve 1.000, P < 0.001). The detail of plasma oxysterols levels and their ratios to total cholesterol are reported for each patient in Tables 1 and 2. The nature of the mutation (truncating versus missense) did not influence the levels of 25-OHC and 27-OHC. Likewise, the four patients with truncating mutations (Patients SAL-930-001, SAL-399-229, SAL-1491-001 and BEL-001) exhibited comparable plasma oxysterol levels compared to patients harbouring missense mutations (Tables 1 and 2). Furthermore, even three siblings (Patient SAL-004) showed quite variable plasma oxysterols levels (Table 2).
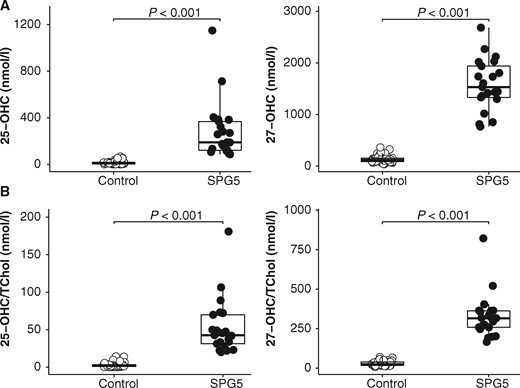
Validation of plasma oxysterols (25-OHC and 27-OHC) as diagnostic biomarkers in SPG5. 25-OHC, 27-OHC (A) and their ratio to total cholesterol (B) are significantly elevated in SPG5 patients compared to controls.
Four controls exhibited 25-OHC levels in the upper normal range, possibly because of the influence of cholesterol levels and fasting on plasma oxysterols. However, none of these healthy individuals exhibited abnormal 27-OHC levels or oxysterols to total cholesterol ratios, emphasizing the relevance to measure both 25-OHC and 27-OHC and their ratios to total cholesterol. In three SPG5 patients, plasma oxysterols were measured repeatedly, at approximately 1-year intervals. Both 25-OHC and 27-OHC remained very high over time, despite some variations in concentrations, while 25-OHC/total cholesterol and 27-OHC/total cholesterol ratios were globally stable over time (data not shown). Oxysterol levels were strictly normal in two patients with cerebrotendinous xanthomatosis (data not shown). Plasma 24S-OHC, not metabolized by the cytochrome P450 7α-hydroxylase, was measured in 20 patients with SPG5 and was found similar (88.3 ± 37.5 nmol/l) to controls (65.7 ± 60.6 nmol/l) (data not shown).
Phase II therapeutic trial in SPG5
All 12 patients completed the 18-month trial. Several adverse events were reported (Supplementary Table 4) but no serious adverse events. Adverse events were mostly unrelated to the treatments except for CDCA with several patients experiencing diarrhoea (Supplementary Table 4), which led to a reduction in the doses of CDCA to 750 mg in eight patients and 500 mg in four patients. Transient muscle pain was reported by three patients under atorvastatin but considered unlikely to be related to the study drug (Supplementary Table 4) as creatine kinase levels remained normal and other concomitant medications or spasticity itself were incriminated instead.
We observed decreased total cholesterol with atorvastatin (P < 0.001, Fig. 4), associated with decreased plasma 27-OHC (P < 0.001, Fig. 4A) and 24S-OHC (P < 0.001, Supplementary Fig. 3) but no significant change in 25-OHC (Supplementary Fig. 3). Plasma 27-OHC to total cholesterol ratio did not change significantly with any of the treatments (Fig. 4A). Plasma oxysterols were not significantly altered by CDCA or resveratrol (Fig. 4A). However, CDCA led to increased levels of total bile acids (P = 0.009, Fig. 4B), associated with increased serum CDCA, LCA and UDCA (P < 0.001, Fig. 4B), and decreased cholic acid and deoxycholic acid (P < 0.001, Fig. 4B). The impact of CDCA on serum bile acids profile was observed even in patients treated with 500 mg daily (data not shown).
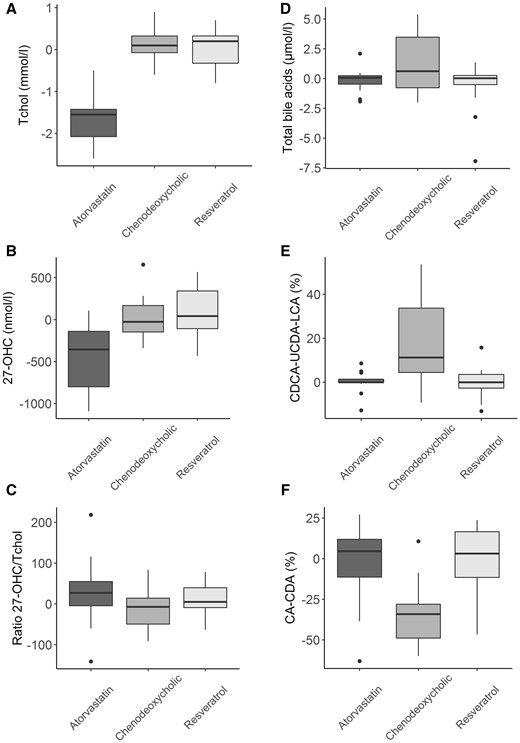
Phase 2 therapeutic trial. Evolution of plasma oxysterols under treatments in SPG5 patients. Atorvastatin led to significantly decreased cholesterol (A) and 27-OHC (B) in SPG5 patients but 27OHC/CT ratio (C) did not change under treatments. Evolution of serum bile acids under treatments in SPG5 patients. Chenodeoxycholic acid led to significantly increased total bile acids (D), serum CDCA and its secondary bile acids LCA and UDCA (E), while CA and its secondary bile acid DCA decreased (F).
Discussion
We validated plasma 25-OHC and 27-OHC as robust diagnostic biomarkers in SPG5 and showed that they shall be used in therapeutic trials evaluating disease-modifying drugs in SPG5. Our short-term trial confirmed that atorvastatin can moderately decrease plasma 27-OHC levels by ∼30%. The confirmation of an abnormal bile acids profile in SPG5 patients, which can be improved by CDCA, also suggests that it may be worth combining atorvastatin (40 mg) and CDCA (500 mg) for the treatment of SPG5 patients. The neurological benefit of these metabolic interventions remains to be evaluated. However, the identification of relevant neurological outcome measures is challenging in patients with long disease duration. Stratification for modifiers of disease progression has been recommended for hereditary spastic paraplegia (Schüle et al., 2016) but may be difficult to apply in small cohorts of patients, like SPG5.
Using our UPLC-MS/MS method with isotopic dilution for oxysterols measurement, we showed that SPG5 patients exhibited an average 19-fold increase in 25-OHC and 11-fold increase in 27-OHC compared to controls. After its first description in a small series of four SPG5 patients (Schüle et al., 2010), only case reports have been presented with plasma 27-OHC measurements (Di Fabio et al., 2014; Vanotti et al., 2014). In our series, plasma oxysterols and their ratios to total cholesterol were increased in all the patients initially detected by molecular screening (n = 14). Conversely, plasma oxysterols proved to be a critical diagnostic tool for SPG5 in patients with unexplained spastic paraplegia (n = 7), subsequently confirmed by CYP7B1 molecular analyses. As previously reported in four SPG5 patients (Schüle et al., 2010), 24S-OHC and its ratio to total cholesterol was not different between SPG5 patients and controls.
Plasma oxysterols ought to be an important biological validation test in SPG5, especially in patients for whom molecular analyses were inconclusive. Likewise, for Patient NIM-001 presenting with a typical SPG5 phenotype but one pathogenic mutation [previously published in Roos et al. (2014)] and one mutation of uncertain significance (amino acid moderately conserved, only one software predicted it as deleterious), increased plasma 25-OHC and 27-OHC levels were determinant to confirm the diagnosis of SPG5. However, the nature of the mutation (truncating versus missense) did not seem to influence the levels of plasma oxysterols, similar to the absence of genotype–phenotype correlation in SPG5. Indeed, the same homozygous truncating mutation (c.1162C>T; p.Arg388*) can be associated either with a severe congenital bile acid synthesis defect (Setchell et al., 1998) or with an adult-onset spastic paraplegia (e.g. Patients SAL-930-001 and SAL-1491-001). While a few patients from our series displayed a moderate increase of plasma 25-OHC compared to controls, 27-OHC values and oxysterols to total cholesterol ratios were consistently and markedly increased in all SPG5 patients. The use of 25-OHC and 27-OHC as ratio to total cholesterol should also circumvent the risk of false positives due to hypercholesterolaemia, or non-fasted samples, since 27-OHC correlates with total cholesterol (Del Puppo et al., 1998). To our knowledge, no other known metabolic defect leads to increased plasma 25-OHC or 27-OHC.
The robustness and specificity of plasma oxysterols in SPG5 supported that they can be used as outcome measures in proof-of-concept therapeutic trials aimed at identifying molecules that can correct the underlying metabolic defect associated with neurodegeneration. Our phase II therapeutic trial showed significantly decreased plasma cholesterol and 27-OHC in SPG5 patients treated by atorvastatin. There was no change in 27-OHC to total cholesterol ratio under treatment, suggesting that the decrease of 27-OHC was only mediated by the decrease of cholesterol. Still, decreasing plasma 27-OHC ought to be beneficial in SPG5 as 27-OHC is thought to be neurotoxic (Wang et al., 2016) and can cross the blood–brain barrier, causing cerebral accumulation, as demonstrated by their high levels in the CSF from SPG5 patients (Schüle et al., 2010). While the relationship between cholesterol and both 27-OHC and 24S-OHC has been established (Thelen et al., 2006), it is not the case for 25-OHC. In fact, 27-OHC is a direct substrate of CYP7B1 (Fig. 1), which may explain why it decreased preferentially with cholesterol-lowering atorvastatin. Likewise, plasma 27-OHC was previously shown to decrease in controls under atorvastatin (Thelen et al., 2006) but not 25-OHC. This may explain why we observed decreased plasma 27-OHC and 24S-OHC levels under atorvastatin but no change in 25-OHC levels.
Furthermore, as suggested before (Schüle et al., 2010), we confirmed that the profile of serum bile acids is abnormal in SPG5 patients with decreased UDCA and LCA, which are secondary bile acids derived from CDCA. As reported in patients with CYP7B1 mutations and severe liver insufficiency, abnormal bile acid levels are likely to be deleterious to the liver (Dai et al., 2014) but their toxicity to the brain remains to be determined. Overall, we observed that total bile acids are decreased in SPG5 patients, with an imbalance between cholic acid and CDCA-derived metabolites (Taoka et al., 2016). This prompted us to test CDCA in patients with SPG5, especially as this is the only drug reported so far to successfully treat severe liver disease related to CYP7B1 mutations (Dai et al., 2014). Our phase II trial showed that the serum bile acids profile from SPG5 patients improved with CDCA, with a restored balance between cholic acid and CDCA pathways as reported in healthy individuals (Nilsell et al., 1983). The absence of changes in oxysterol levels in SPG5 patients receiving CDCA may be due to the insufficient treatment duration (2 months) since plasma cholestanol may take up to 6 months to normalize in cerebrotendinous xanthomatosis patients treated by CDCA (unpublished results). Likewise, decreased oxysterols levels were reported several years after treatment with CDCA in the CYP7B1 mutated child with severe cholestatic disease (Dai et al., 2014). Unlike patients with cerebrotendinous xanthomatosis whose diarrhoea usually resolves upon treatment with CDCA 750 mg daily (Nie et al., 2014), SPG5 patients tended to experience diarrhoea with 1000 mg daily of CDCA. Doses between 500 mg and 750 mg were better tolerated and our data suggest that 500 mg may be sufficient for long-term effect on these metabolic pathways.
Five patients presented here were partially reported previously (Goizet et al., 2009). This large series of 21 patients confirmed the clinical phenotype of SPG5 characterized by a relatively pure form of spastic paraplegia associated with prominent posterior column sensory impairment (decreased or abolished vibration sense) and mild-to-severe bladder dysfunction. Delayed somatosensory evoked potential and normal peripheral nerve conduction was consistent with pre-ganglionary or medullary sensory involvement (Manganelli et al., 2011; Vanotti et al., 2014). In addition, brain MRI showed diffused posterior white matter involvement (67%), a finding otherwise common in patients with spastic paraplegia and cortico-spinal degeneration of genetic or metabolic origin (de Souza et al., 2017). Spinal MRI may be normal or showing a variable degree of atrophy. These clinical features were quite uniform in all our SPG5 patients but not specific. Hence, phase III therapeutic trials in SPG5 await clinical, imaging and/or electrophysiological outcome measures with sufficient effect sizes. Considering the rarity of the disease, an international effort is probably mandatory in that regard, including for the evaluation of putative novel readouts such as diffusion tensor imaging or magnetic resonance spectroscopy of the spine, as suggested for other diseases with prominent medullary involvement like adrenomyeloneuropathy (Castellano et al., 2016).
In conclusion, we strongly recommend measuring plasma oxysterols as a first-line investigation in any patient with an unexplained progressive spastic paraplegia with sporadic or recessive inheritance, in order to guide genetic analyses. Of note, SPG5 is not so rare and may account for up to 3% of HSP patients with a mutation identified on next generation sequencing panels (unpublished results). Our short-term phase II therapeutic trial suggests that atorvastatin and CDCA may exert complementary actions on the metabolic alterations observed in SPG5 patients. However, these drugs may not be benign in the long term and the identification of relevant neurological readouts remains critical to assess the benefit of these metabolic interventions in SPG5 on the CNS before a chronic administration can be proposed.
Acknowledgements
We wish to thank warmly all the patients participating to these studies. The authors also wish to thank Pr Koenig and Mrs Larrieu for molecular analysis of Patient MON-001, Dr Coutelier for molecular analysis of Patient BEL-001, and Dr Petiot and Dr Ossama for sharing clinical information.
Funding
The biomarkers study was funded by the Agence Nationale de la Recherche (ANR) (Project 2010BLAN112601/LIGENAX) (project E-Rare ANR 2013-2018/Neurolipid), the Association Française contre les Myopathies (AFM) (14879/MNM2 2012), the Conseil Régional d’Aquitaine (CRA) (2011-0151/LIGENAX), the Association Strumpell-Lorrain (ASL) (2011-0135), and the Pôle de compétitivité Prod’Innov. The therapeutic trial was funded by the Association Strumpell-Lorrain (ASL) (2014-2017), the Carnot Institute, Inserm (COSSEC) and the FP7 HEALTH-Neuromics project.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- CDCA
chenodeoxycholic acid
- LCA
lithocholic acid
- OHC
hydroxycholesterol
- SPG5
spastic paraplegia type 5
- UDCA
ursodeoxycholic acid
- UPLC-MS/MS
ultra performance liquid chromatography-tandem mass spectrometry
References
Del Puppo M, Kienle MG, Petroni ML, Crosignani A, Podda M.
Roos P, Svenstrup K, Danielsen ER, Thomsen C, Nielsen JE.
Schüle R, Brandt E, Karle KN, Tsaousidou M, Klebe S, Klimpe S, et al.
Siam A, Brancale A, Simons C.
Author notes
Cecilia Marelli and Foudil Lamari equally contributed to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}