Article Text

Abstract
Coeliac disease is a chronic immune-mediated disorder that primarily affects the gastrointestinal tract. There is an inflammatory response in the intestine to the ingestion of gluten which improves with a gluten-free diet. Many patients, especially adults, may be asymptomatic or have only extraintestinal symptoms at onset without any of the classical coeliac symptoms. In the last two decades there have been increasing numbers of reports describing neurological complications of coeliac disease, especially ataxia, peripheral neuropathy and epilepsy. This literature has become quite controversial, with disputes over the definition of coeliac disease and gluten sensitivity, whether neurological complications are caused by coeliac disease or are epiphenomena, and whether the proposed complications respond to a gluten-free diet. This review uses an evidence-based approach to critically assess this literature and provides guidelines for the evaluation and management of these patients.
Statistics from Altmetric.com

In the last two decades, there have been increasing numbers of papers describing neurological complications of coeliac disease. Indeed, this has become a rather contentious literature, at times provoking personal attacks between authors. Disputed issues include the definition of coeliac disease and “gluten sensitivity”, whether certain complications are caused by coeliac disease or are epiphenomena, and whether any of these proposed complications respond to a gluten-free diet. The purpose of this review is to use an evidence-based approach to critically assess this controversial literature.
DEFINITION
Coeliac disease is a chronic immune-mediated inflammatory disease which develops in genetically predisposed individuals. There is a T-cell mediated immune response to ingested gluten and related proteins found in wheat, rye and barley. This response produces inflammation, villous atrophy and crypt hyperplasia in the proximal part of the intestine. It is more common in people who are HLA DQ2 and DQ8 positive.1 It is associated with several other disorders (table 1).
CLINICAL MANIFESTATIONS
The clinical manifestations vary greatly and although coeliac disease was once perceived to be primarily a paediatric disorder, the diagnosis is increasingly being made in adult life (unfortunately many adults are still misdiagnosed as having irritable bowel syndrome). The classic presentation, in children and adults, includes steatorrhoea, vomiting, abdominal pain, diarrhoea, muscle wasting, anaemia and nutritional deficiencies. In less typical forms, both in children and adults, gastrointestinal symptoms are less prominent or even absent, and extraintestinal features (table 2) are what bring many patients to medical attention. On the other hand, some individuals have “asymptomatic” or “silent” coeliac disease without any classical or even atypical symptoms, but are diagnosed after incidental intestinal biopsy or positive serology.1–3
EPIDEMIOLOGY
The true prevalence of coeliac disease is unknown due to the increasing recognition that a high percentage remain undiagnosed. A common expression in the coeliac literature is the “coeliac iceberg”. Clinically manifest coeliac disease is the peak, and the remaining patients are below the surface of the sea (fig 1). One estimate is that the total prevalence may be 1 in 100, making this a common disorder, although many of these patients are asymptomatic.2 4 There is geographic variation—it is more common in individuals of European descent, lower in India, South America, the Middle East, Africa and Asia.

PATHOPHYSIOLOGY
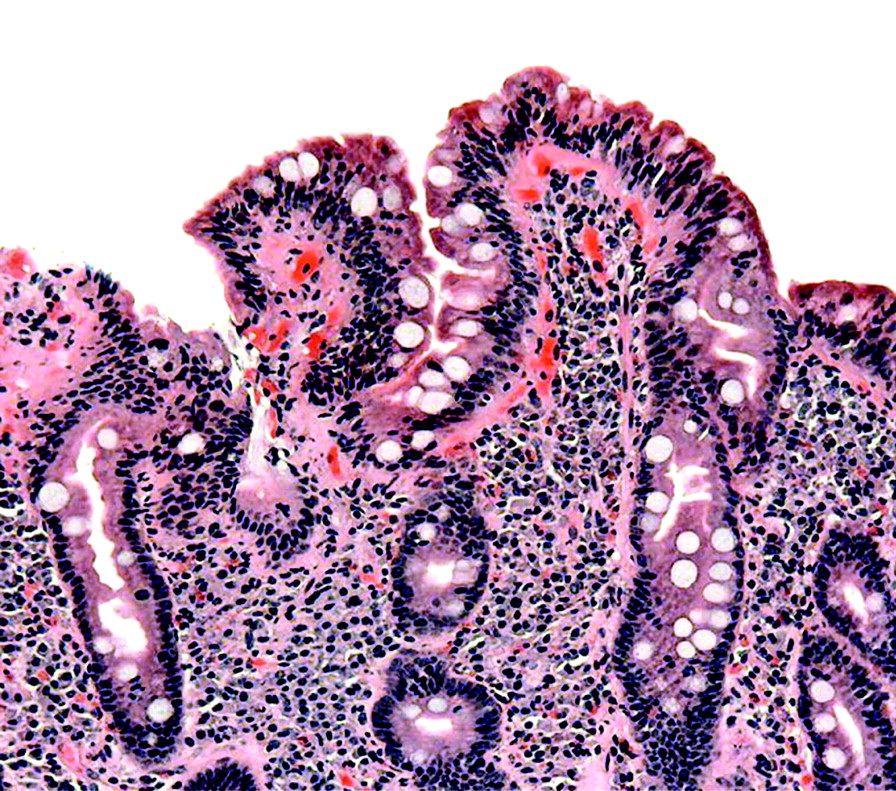
Gluten is mainly found in wheat, rye and barley. When gluten is ingested, it is broken down into peptides, particularly gliadin. It is thought that gliadin binds to antigen-presenting cells expressing HLA-DQ2 and HLA-DQ8. This is facilitated by the enzyme “tissue transglutaminase”. The peptides are then presented to intestinal mucosal T-lymphocytes which become activated, producing antibodies and cytokines, resulting in inflammation and intestinal mucosal injury. Small bowel biopsy shows variable amounts of villous atrophy, crypt hyperplasia and inflammation (fig 2). Treatment with a gluten-free diet results in improvement in intestinal structure and symptoms.5
DIAGNOSIS
At the present time, serum IgA EMA (endomysial antibody), IgA tTG (tissue transglutaminase antibody) and small bowel biopsy are the accepted tests to diagnose “classical” coeliac disease. The sensitivity and specificity of the serological tests for coeliac disease are based on the diagnostic gold standard of intestinal biopsy. The histological appearances show a progression of changes beginning with an increase in epithelial lymphocytes, but this is not specific for coeliac disease. Next enlarged crypts are seen, followed by variable degrees of villous atrophy. The serological tests are most sensitive with more severe villous atrophy. However, there is interobserver variability in pathological interpretation of biopsies at all levels of severity, most apparent with intermediate stages of villous atrophy.6 In addition, there are patients with positive serology but only minimal or no histological changes at all who may become symptomatic later in life (latent coeliac disease). Thus, the “gold standard” may be an imperfect standard.
Antigliadin antibodies (AGA) have the lowest sensitivity and specificity compared with intestinal biopsy, although they may be helpful for assessing compliance with a gluten-free diet. Both IgA EMA and IgA tTG are based on the target antigen tTG, while IgA and IgG AGA are based on the target antigen gliadin. A recent review7 found that:
IgA EMA had a sensitivity of 90–97% and specificity close to 100%;
IgA tTG a sensitivity of 90–98% and specificity between 95–99%;
IgA AGA a sensitivity of 75–95% and a specificity of 80–90%;
IgG AGA a sensitivity of 17–100% (wide variation between studies) and a specificity of 70–80%.
Variations are partly due to differences in assay techniques, cut-off points, and histological interpretations. Although some studies have shown differing results,8 most have shown high sensitivity and specificity for IgA EMA and IgA tTG when using a positive intestinal biopsy as the gold standard. Because most of the studies were performed in tertiary care centres, where the prevalence of coeliac disease is greater than the general population, the predictive values may be different when these tests are applied to the practising neurologist’s patient population. It is important that neurologists are familiar with the sensitivity and specificity of the assays used by their own local laboratories, and the populations used as reference populations.
TREATMENT OF COELIAC DISEASE
A gluten-free diet is the mainstay of treatment. After starting the diet, patients should improve in a few weeks, some within 48 hours. However, biopsy changes may take 2–3 months to improve, but may not return to normal. Both IgA tTG and IgA AGA levels decrease in the months following institution of a gluten-free diet, and are useful for monitoring dietary compliance. Rarely, glucocorticoids are used for refractory patients with severe disease. Patients with severe disease also receive supplements to correct nutritional deficiencies caused by malabsorption.5
NEUROLOGICAL COMPLICATIONS
Malabsorption is a well-described complication of coeliac disease and there are easily understandable neurological complications due to vitamin B12 deficiency (myelopathy, neuropathy), vitamin D malabsorption (myopathy), and vitamin E deficiency (cerebellar ataxia and myopathy). Malabsorption complications are now less frequent. Other complications are more controversial, and have been reported in patients without malabsorption.
GLUTEN ATAXIA
Association between ataxia and gluten antibodies
Ataxia has been reported in some coeliac disease patients in many studies. The first were primarily case reports and case series, followed by case-control studies. Hadjivassiliou et al9 described 28 patients, all with gait ataxia, most with limb ataxia as well. They were described as having “gluten sensitivity” on the basis of increased titres of AGA. Eleven patients had typical biopsy-positive coeliac disease, six had cerebellar atrophy on MRI, and autopsies on two patients revealed lymphocytic infiltration of the cerebellum. They proposed the term “gluten ataxia” for patients with AGA who might not have enteropathy or other coeliac antibodies. Previously the same authors had found that 57% of patients with “neurological dysfunction of unknown cause” had increased titres of AGA versus 12% of healthy blood donors.10 They also studied 268 patients with ataxia and compared them to 1200 normal controls;11 41% of idiopathic ataxia, 14% of hereditary ataxia, 15% of multiple system atrophy patients and 12% of controls had increased AGA.
Several other groups have reported an increased frequency of “gluten sensitivity” in patients with idiopathic cerebellar ataxia:
Pellecchia et al12 found 3 of 24 patients with ataxia of unknown aetiology had coeliac disease (AGA and biopsy positive), with none of 23 patients in a control group consisting of ataxic patients with known diagnoses (autosomal dominant cerebellar ataxia and Friedreich’s ataxia).
Burk et al13 reported 12 of 104 patients with ataxia and “gluten sensitivity” defined by positive antibodies only. Just two of these patients had positive biopsies.
Bushara et al14 found 7/26 (27%) patients with sporadic ataxia and 9/24 (37%) of autosomal dominant ataxias had increased AGA titres. There was no control group.
These studies claimed that there was a greater percentage of ataxic patients who had coeliac disease or gluten sensitivity than control groups. In most of these reports, IgG AGA levels were used to screen for “gluten sensitivity”.
On the other hand, there are studies which report the opposite—that there is no relation between coeliac disease or AGA and ataxia. Abele et al studied sporadic ataxia, recessive ataxia, and dominant ataxia and found that positive antibodies were no greater than in controls.15 However, there was a non-statistically significant trend in ataxic patients and the sample size was small. Combarras et al studied 32 patients with sporadic ataxia and found none with positive antibodies, but again the sample size was small.16 Wong studied 56 ataxic patients and 59 controls and found 6/56 ataxia patients were positive for either IgG or IgA AGA compared with 5/59 controls, representing no significant statistical association between AGA and ataxia.17 Lock et al reported similar findings, but again had a small sample size.18
Treatment of ataxia
Hadjivassilou et al evaluated dietary treatment in a non-randomised cohort study of patients with sporadic ataxia with positive AGA, with or without enteropathy.19 Patients and examiners were not blinded, though quantitative assessments of ataxia were performed. The controls were 14 patients who refused the diet. They reported improvement in ataxia in the treatment group.
How to evaluate these studies and others like them
Practical neurologists must feel comfortable applying certain criteria to each article to assess any sources of bias, especially when the data are conflicting (tables 3 and 4) Each of the studies described above, both for and against the hypothesis, had one or more methodological flaws that created bias:
Studies that draw patients and controls primarily from tertiary care centres (narrow spectrum) usually demonstrate referral bias. The prevalence of many diseases is greater in a referral centre than in the patients who appear in the office of the general neurologist. There are limited data on the prevalence in a large general population but Ludvigsson et al did do a retrospective population based study of 14 000 patients with coeliac disease using Swedish national registers, and found some association with peripheral neuropathy, but not with ataxia (neither sporadic nor hereditary), Parkinson’s disease, Huntington’s, myasthenia, spinal muscular atrophy or multiple sclerosis.20 They found prior peripheral neuropathy was associated with subsequent coeliac disease. Retrospective registry studies have case ascertainment inaccuracies, but at least are a start in getting data as they apply to general populations.
Almost all the studies were retrospective, with a narrow spectrum of patients and controls, when controls were used at all.
In most studies, independent evaluators did not measure or characterise the disease outcome (for example, ataxia), with other evaluators assessing the risk factor (coeliac disease) or gluten sensitivity.
Independent and blinded assessments should be done even for pathological interpretation of biopsies, but they were not.
Different studies often used different “gold standards” for case definition of coeliac disease and gluten sensitivity.
The studies that attempted to disprove the association had inadequate sample sizes.
Thus, the differences between these studies may be due to case ascertainment differences, referral bias, inadequate sample size, lack of prevalence data in age-matched general populations, lack of blinding, differences in spectrum of patients, and differences in assay techniques, cut-off values and histological interpretation.
Bottom line
At this time, the evidence is conflicting. From a purely evidence-based analysis of methodology, it can only be said that it is possible there is an association between coeliac disease or AGA and ataxia, but this association would need to be confirmed by a wide range of investigators doing better designed studies.
Antibodies may directly cause a disease, be unrelated to the disease, or be a covariate (associated with some other process that may cause the disease). Do AGA or other coeliac auto-antibodies by themselves cause neurological disease, or are they merely a surrogate marker for another disease-causing process? It is notable that AGA have been reported in hereditary ataxias and other inherited disorders:
Shill et al found 9 of 12 patients with hereditary cerebellar degeneration were positive for antiganglioside antibodies and 7 of these 9 had “gluten sensitivity” as well.21 All three multiple system atrophy patients had both antiganglioside antibodies and “gluten sensitivity”. Antiganglioside antibodies have been associated with acquired cerebellar degeneration and peripheral neuropathy. The coexistence of coeliac antibodies and antiganglioside antibodies in both familial and idiopathic ataxias raises the possibility that these antibodies, and perhaps other as yet undiscovered antibodies to cerebellar or other neuronal antigens, may also be related to some other primary pathogenic process, perhaps related to HLA-type or other genetic factors. However, it should be noted that antiganglioside antibodies have been found in coeliac disease patients both with and without neurological disorders.
Bushara et al studied 52 patients with Huntington’s disease and found that 44% had positive AGA.22
Finally, 10–20% of individuals from the general population may have AGA and stay asymptomatic.
There have been no systematic studies of the possible relation of cell-mediated immunity abnormalities and neurological problems in coeliac disease patients.
PERIPHERAL NEUROPATHY
Peripheral neuropathy in association with coeliac disease has also been described, but again the data are poor and conflicting.
Chin et al found 20 patients with coeliac disease in a retrospective chart review23; some had known coeliac disease when they were referred to their neuropathy clinic, others were diagnosed with coeliac disease during evaluation of their neuropathies. However, their institution had both a peripheral neuropathy and a coeliac disease centre making referral bias rather likely. Patients presented with burning, tingling and numbness in hands and feet, with distal sensory loss on examination, frequently without any gastrointestinal symptoms. EMGs were either normal or mildly abnormal. Sural biopsy in three patients showed mild to severe axonopathy. They noted antiganglioside antibodies in 65% of these patients in addition to coeliac antibodies and positive intestinal biopsies. In addition, coeliac disease was noted in 2.5% of all their neuropathy patients, diagnosed with serology and small bowel biopsy. Two of six patients placed on a gluten-free diet had “subjective improvement” in symptoms, but not on objective measures. Intravenous immunoglobulin (IVIg) was tried in five patients, with no improvement in four. Chin et al also reported six patients with multifocal axonal polyneuropathy with biopsy proven coeliac disease; in five the neurological symptoms antedated the diagnosis of coeliac disease by as much as four years, in the sixth case, the patient had neurological symptoms beginning at age 37 with coeliac disease diagnosed at age 79.24
Brannagan et al reported eight coeliac patients with sensory neuropathies.25 Coeliac disease was diagnosed in five patients (biopsy) after the neuropathy began, one as an infant, with the neuropathy developing in the sixth decade. Four patients reported symptomatic improvement on a gluten-free diet, one patient no improvement. In two patients, the symptoms appeared while already on a gluten-free diet. This was clearly an uncontrolled case series which provides low level evidence.
De Sousa et al studied 62 patients with clinical sensory neuropathy and abnormal skin biopsies (reduced epidermal nerve fibre density or morphological changes in epidermal nerve fibres consistent with small fibre neuropathy).26 Eleven patients had abnormal tTG, antigliadin IgA and IgG antibodies, but normal duodenal biopsy. Six patients had biopsy proven coeliac disease. The authors admit the high percentage of coeliac patients may represent referral bias.
The opposite hypothesis—no association between neuropathy and coeliac disease—has been suggested by others. Rosenberg et al retrospectively reviewed patients with peripheral neuropathy in their own centre, excluding patients with ataxia and symptoms suggesting small fibre neuropathy.27 They included only patients with biopsy proven coeliac disease, and investigated 53 patients with no cause for their neuropathy. Of these, 12 patients had already died and three had been lost to follow-up before diagnoses could be made. In 22 cases, they discovered other causes of the neuropathy. Of the 16 left, one had increased AGA but negative intestinal biopsy. This was retrospective, excluded patients with ataxia (who may have had coeliac disease), excluded small fibre neuropathy patients (who may also have had coeliac disease), and they could not investigate the 15 who had died or were lost to follow-up.
Dietary treatment of neuropathy
Hadjivassiliou et al describe a non-randomised trial of 25 neuropathy patients with gluten sensitivity defined by positive AGA with or without EMA or tTG anitbodies.28 The control group was 10 patients who refused the diet. The outcome was mean change in sural nerve action potential amplitude after one year. The secondary outcome was a change in a composite score combining multiple neurophysiological variables; there was no difference in score between patients and controls. The authors reported that the group in the intention-to-treat arm had “significant” improvement in neuropathy, measured by another parameter, the sural sensory amplitude. However, a single patient in the treatment group had an improvement from 1.5 to 5.8 microvolts, probably having an inappropriately large impact statistically on the mean calculations, although the data were not provided in enough detail. In addition, the authors noted that if a comparison is done between a treatment group with complete elimination of AGA (compliant) with a group consisting of those who refused the diet plus those who were on the diet but did not eliminate AGA (non-compliant), there was no significant difference. This suggests that effective treatment did not alter the outcome. In fact, the mean differences in sural sensory amplitudes fell within the usual technical variation from study to study due to small variations in limb temperature, electrode placement, and normal small measurement differences encountered from session to session.
Tursi et al described no improvement in neuropathy with diet, but used an inadequate definition of neuropathy.29 Rigamonti et al report two patients with predominantly motor neuropathy who “improved” with a gluten-free diet (an anecdotal study).30
Bottom line
Most of the studies are case series, providing the lowest level of evidence. The case-control studies and therapeutic trials had one or more of the same methodological problems described in the ataxia literature. There are no prospective studies, nor adequate control groups, and there has been a lack of blinding of examiners, use of “volunteer subjects” (choosing as treatment participants only those who agree to therapy, using those who refuse therapy as controls), narrow spectrum (referral centres), variable standards for case definition of coeliac disease, gluten sensitivity, and peripheral neuropathy, and variable serological assays. Electrophysiology was not always done, and when it was did not always conform to current research definitions for peripheral neuropathy. Again, based on principles of evidence-based medicine and evaluations of methodology, there is only a “possible” association due to lower levels of evidence and conflicting evidence. There is not yet convincing evidence of causality.
EPILEPSY
There have been studies that have reported an association between epilepsy and coeliac disease:
Chapman et al described 165 patients with coeliac disease (biopsy positive) and a control group of 165 age- and sex-matched controls from a general practice.31 Nine of the coeliac group had epilepsy (seven of them temporal lobe epilepsy), with no cases in the control group and a national prevalence of 0.5% obtained from the literature.
Cronin et al found an increased prevalence of coeliac disease in patients, attending a seizure clinic in Ireland compared to a control group consisting only of pregnant women.32 This control group was inappropriate because coeliac disease is associated with infertility, making the control group biased against coeliac disease.
Luostarinen et al did a retrospective records review of 900 epilepsy patients, and the occurrence of coeliac disease in those with epilepsy of unknown aetiology (199 patients) was investigated, based on serology and intestinal biopsy.33 Five of the 199 patients had a previous diagnosis of coeliac disease. The prevalence of coeliac disease was 2.5% compared to “local prevalence” of disease of 0.27%.
Mavroudi et al studied 255 children in Greece with idiopathic epilepsy attending a paediatric neurology outpatient clinic at a university-associated hospital (referral bias).34 The diagnosis of coeliac disease was made by serology plus positive intestinal biopsy. The control group was 280 healthy children from a paediatric outpatient clinic who came for routine health evaluation. IgG AGA were found in 35 patients and 56 controls, while IgA AGA were found in five patients but no controls. Two patients had positive anti-tTG, none in controls. No children had positive anti-EMA. Biopsies were done only on children with positive IgA AGA (total of five), two were positive. There were thus just two patients with confirmed coeliac disease, none of the controls.
Dalgic et al studied Turkish children and found two of 70 epilepsy patients had coeliac disease versus none in 103 controls (children admitted to the hospital for other reasons).35 The diagnosis was made by anti-tTG antibody screening followed by intestinal biopsy.
As expected in this literature, there are other studies finding no association:
Dayangku et al studied 801 coeliac patients and found no significant statistical difference between their prevalence of epilepsy versus controls.36 Twenty one coeliac patients had a lifetime history of seizures, but only nine had active epilepsy, representing 1.1% of coeliac patients. This was compared to a general estimate of the prevalence of epilepsy of 0.5–1% derived from the literature. There was no figure for prevalence of epilepsy in a suitable population control group. They also found no temporal relation between the development of coeliac disease and epilepsy. It was not clear how many of the coeliac patients were on gluten-free diets and how many were untreated. It is possible that seizures may be more frequent in non-treated or non-compliant patients. One strength of this study is that the patients were drawn from a general catchment area, not a tertiary care centre.
Pratesi et al studied 255 epileptic patients from epilepsy clinics (likely referral bias) and compared them to 4405 patients who were having routine blood testing.37 Coeliac disease was diagnosed by IgA EMA assay and intestinal biopsy. The prevalence of coeliac disease was 7.84 per 1000 in the epileptic patients and 3.41 in the controls, which was not statistically significant, although the authors described their results as “suggestive” of an increased prevalence.
Ranua et al studied a cohort of 968 Finnish patients with all types of epilepsy and matched controls from the general population, looking at the frequency of IgG and IgA AGA, IgA tissue transglutaminase antibodies, and IgA anti-endomysial antibodies.38 The prevalence of antibodies was no different in the patients and controls. However, they did find that IgA AGA was more frequent in patients with primary generalised epilepsy than controls.
There are studies that have reported a specific rare syndrome—bilateral occipital calcifications and seizures in patients with coeliac disease, especially in Italy, Spain and Argentina (fig 3).39 There has been some speculation that this may be a genetic syndrome with geographic specificity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dietary treatment
Gluten restriction in some studies has resulted in a reduction of seizure frequency with a decrease in antiepileptic medication needed to control intractable seizures. These are primarily case reports, without systematic study. There have been no prospective controlled trials studying the effect of gluten-free diet in patients with coeliac disease who also have epilepsy.
Bottom line
There are the same methodological issues in the epilepsy literature as there were in the ataxia and neuropathy literature already discussed. From the evidence-based perspective, there is conflicting evidence whether there is or is not an association between coeliac disease or auto-antibodies and epilepsy. As yet there is no compelling evidence that there is a causal relation. There probably is a specific syndrome—coeliac disease with epilepsy and calcifications—which is rare and perhaps geographically specific.
MISCELLANEOUS ASSOCIATIONS
There are other neurological disorders that have been reported in association with coeliac disease or coeliac antibodies. These are in the form of case reports and case series, and do not represent high levels of evidence. They include myopathy, autonomic neuropathy, white matter lesions, headache, cognitive impairment, multiple system atrophy, dystonia and childhood stroke.
UNRESOLVED ISSUES
What is gluten sensitivity and what does it mean for patients? In the past coeliac disease has been defined by serological abnormalities plus abnormal intestinal biopsy. Some of the studies noted above have proposed that elevated AGA represent “gluten sensitivity”, itself possibly representing a disease state, or at least a risk factor for the associated neurological conditions.
A related issue is the sensitivities and specificities of diagnostic tests, both of which are a function of some diagnostic “gold standard” for the disease in question. Without this gold standard, the concepts of true positive, false positive, true negative and false negative have no meaning. If AGA represent a disease state or potential disease state separate from classical coeliac disease, how do we define when this is a disorder, because 10–20% of healthy individuals may have antibodies and no obvious clinical consequences?
Higher levels of evidence are needed to establish associations between coeliac disease, auto-antibodies and neurological disease. And even when an association becomes strong, there are other factors which should be considered before believing that the association represents a causal relation (table 5).
“TESTS” OF CAUSALITY AS APPLIED TO COELIAC DISEASE AND NEUROLOGICAL DISORDERS
Strength of association
The higher the level of evidence, the more likely there is an association, and the higher the odds ratios or relative risk ratios, the stronger the association is likely to be. This category also includes specificity of association; what other disorders can create the same outcome? For example, diabetes and thyroid disease (both comorbidities of coeliac disease) can be complicated by peripheral neuropathy. Also, how many patients with the “risk factor” (that is, coeliac antibodies) do not get the outcome of interest (for example, ataxia)?
Consistency
Is there a repeated consistent association using different methodological designs, different investigators and different settings? In the studies discussed above, there have been significant variations.
Temporal relation
Exposure to the risk factor (coeliac disease or antibodies) should lead within a relatively consistent time period to the outcome being measured (for example, ataxia). This is difficult to ascertain in coeliac disease or gluten sensitivity because so many patients are asymptomatic, and so it is difficult to say when disease onset occurred. Some patients in the studies noted above had a 40–50 years lapse between onset of coeliac disease and neurological complications, calling into question causality. Retrospective studies in particular have difficulty noting the exact time of onset of disease.
Is there a “dose-response” gradient?
In the coeliac studies there has been no consistent relation between any markers of disease such as intestinal biopsy changes, antibody titres, traditional coeliac symptoms and neurological complications. This is not necessary to prove causality, but would lend credence to the concept.
Relation to treatment
Does treatment and improvement of the underlying disorder correlate with improvement in the measured outcome (ataxia, neuropathy, seizures)? Ideally this should be determined by a randomised controlled trial with blinding of patient and examiner. However, there are circumstances in which comparison to placebo may not be ethical. In this case it may be inappropriate not to treat patients with coeliac disease who are symptomatic in other ways, because the treatment is only a change in diet, it is effective for other coeliac symptoms, and is an intervention which is non-invasive and does not produce significant harm. Several studies have used non-treated patients who have refused treatment or who are non-compliant with their diets as control groups. The use of “volunteer subjects”, selecting as treatment subjects only those who are willing to go on the diet while unwilling treatment participants are used as controls, is known to introduce bias, but may be the best that can be achieved in this case. If this is accepted, effort has to be made to ensure the sample size is adequate, and that the outcomes are evaluated by blinded examiners, although the patients themselves are not blinded. The effect of non-blinded examiners has been underestimated in the studies discussed above. Other problems in assessing the results of treatment include the difficulty patients have in maintaining a gluten-free diet and the possibility that gluten withdrawal may not improve established neurological disease.
Biological plausibility
There are other autoimmune disorders, such as systemic lupus erythematosus, Sjögren’s syndrome and rheumatoid arthritis that have neurological complications, sometimes pre-dating the symptoms and diagnosis of the primary disease. It is entirely plausible that neurological complications can occur as an autoimmune phenomenon due to coeliac disease. Although coeliac disease itself is primarily T-cell mediated, there may be auto-antibodies which could cross-react with cerebellar antigens, for example, as has been proposed. There are also studies which have begun to further investigate the relation of antiganglioside antibodies to coeliac disease and neurological complications.
Have alternative explanations been explored?
This includes scenarios such as chance association of the outcome (for example, ataxia) with the risk factor (coeliac disease or gluten sensitivity). The prevalence of coeliac disease and its related antibodies is likely higher than previously suspected in general populations. This category also includes the possibility that coeliac disease or the antibodies are a covariate, related to some other process that is actually causing disease.
Causality and antibody mediated disease
Criteria have been proposed that would suggest that antibodies mediate a specific neurological disease:40
Autoantibodies should be present in the serum or cerebrospinal fluid of most patients with the condition and not in healthy patients or other control patients with other unrelated disorders.
Autoantibodies should be demonstrated to bind to target antigens at sites of pathology that are specific to patients and not in normal controls.
Plasma exchange should have a therapeutic effect
Injection of serum or immunoglobulin should transfer the disease to experimental animals.
An equivalent disease should be induced in animals by sensitising susceptible animals with the target antigen.
There has been some work related to these criteria, but not yet enough to satisfy causality.
WHAT DO WE DO NOW?
While we wait for more definitive studies to be done, what should be the strategy of the practical neurologist? The everyday practise of medicine requires weighing costs and benefits for each individual patient under conditions of uncertainty, with inadequate data. When faced with these dilemmas, we often consider most the concept of harm. We are more willing to propose a therapy with lower degrees of evidence of therapeutic efficacy when the severity of the neurological illness produces significant pain or disability, and the potential harm of the treatment is low. Therefore, when faced with a patient who has progressive disability due to ataxia or neuropathy of unknown aetiology or uncontrolled seizures, it is reasonable to discuss with the patient the inadequacy of our knowledge, but offer evaluation for coeliac disease, including anti-tTG, anti-EMA and AGA, as well as a confirmatory intestinal biopsy if the antibodies are positive.
Dietary treatment can be offered to those with either classical coeliac disease (with positive intestinal biopsy) or those with positive serology alone, as long as the patient understands we have no definitive clinical trial data that a gluten-free diet will improve neurological function either in patients with classical coeliac disease or those with just positive serology.
In those patients with positive intestinal biopsies who do not improve neurologically, a gluten-free diet should still be recommended to reduce the chance of other coeliac complications in the future including intestinal lymphoma.
In those patients with positive serology but negative intestinal biopsy who do not improve neurologically, several strategies might be reasonable. Some of these patients may truly be “false positives” who will never develop any coeliac symptoms or other coeliac complications in the future, may have neurological disorders due to unrelated disease, and may not benefit from a gluten-free diet. However, others in this group may benefit from continued gluten-free diet. This would include those patients who have had negative biopsies due to patchy involvement of the intestine, variability in biopsy interpretation, or who have “latent coeliac disease” (positive serology and negative intestinal biopsy with the potential to develop intestinal changes and other coeliac complications in the future). Thus, choices for the group with negative intestinal biopsy could include serial monitoring of serology (rare patients have spontaneous reversion to normal serology), repeat small bowel biopsy, or a continued trial on a gluten-free diet. Individualised informed discussions are necessary, but in the end it may be difficult to determine to which of the above categories in this group a given individual will belong, and maintenance of the diet may be the most prudent strategy until further data are available.
There are those who may feel that this overall approach will lead to unnecessary testing and too many “false positives”. This type of strategy is acceptable only when the consequences of the neurological illness are significant, the risk of any intervention low and there may be some chance, even remote, that the patient may benefit from treatment. This equation changes when the risk of intervention is greater, such as plasma exchange, IVIg, or immunosuppressive therapy; these interventions should only be considered within the context of randomised trials until more data become available.
Practice points
Do not assume that all published papers have equal validity, even if they are in the New England Journal of Medicine or the Lancet.
Read the methods sections of studies carefully.
Assess bias and level of evidence (tables 3 and 4).
Assess proof of causality (table 5).
Have any treatment studies been well-controlled trials, with patients similar to your own?
It is possible that coeliac disease and antibodies are associated with neurological complications, but at this time the evidence is poor, conflicting, and causality has not been demonstrated.
An evaluation for coeliac disease and antigliadin antibodies in patients with progressive ataxia, neuropathy and uncontrolled seizures of unknown origin can still be justified if patients are informed of the inadequate data on causality and treatment benefit. This strategy is acceptable only because the risks of diagnosis and treatment—gluten exclusion—are low.
Other therapies with greater chance of harm should be offered only within randomised trials with appropriate informed consent.
The strategies above are temporary until more data are available; follow the literature, carefully and critically.
Acknowledgments
This article was reviewed by Graeme Hankey, Perth, Australia.
REFERENCES
Other content recommended for you
- Neurological complications of coeliac disease
- Neuropathy associated with gluten sensitivity
- Gluten sensitivity masquerading as systemic lupus erythematosus
- Hippocampal sclerosis in refractory temporal lobe epilepsy is associated with gluten sensitivity
- Celiac disease and non-celiac gluten sensitivity
- IgG1 antiendomysium and IgG antitissue transglutaminase (anti-tTG) antibodies in coeliac patients with selective IgA deficiency
- Gluten sensitivity as a neurological illness
- Coeliac disease in Williams syndrome
- A prospective study of the prevalence of undiagnosed coeliac disease in laboratory defined iron and folate deficiency
- Autonomic neuropathy and coeliac disease