Article Text

Abstract
Metabolic myopathies are a diverse group of rare genetic disorders and their associated muscle symptoms may be subtle. Patients may present with indolent myopathic features, exercise intolerance or recurrent rhabdomyolysis. Diagnostic delays are common and clinicians need a high index of suspicion to recognise and differentiate metabolic myopathies from other conditions that present in a similar fashion. Standard laboratory tests may be normal or non-specific, particularly between symptomatic episodes. Targeted enzyme activity measurement and next-generation genetic sequencing are increasingly used. There are now specific enzyme replacement therapies available, and other metabolic strategies and gene therapies are undergoing clinical trials. Here, we discuss our approach to the adult patient with suspected metabolic myopathy. We outline key features in the history and examination and discuss some mimics of metabolic myopathies. We highlight some disorders of glycogen and fatty acid utilisation that present in adulthood and outline current recommendations on management.
- metabolic disease
- muscle disease
- neuropathology, muscle
- myopathy
Statistics from Altmetric.com
Introduction
The metabolic myopathies are a heterogeneous group of genetically determined conditions that may present at any age (box 1). They are characterised by defects in the biochemical pathways of storage, mobilisation and utilisation of the substrates of muscle energy generation. Skeletal, cardiac and smooth muscle dysfunction, as well as brain and endocrine involvement, contribute to their protean manifestations. Importantly, while many patients have had symptoms since childhood, they often do not come to the attention of a physician until later in life.
Metabolic myopathies, focussing on those that may present in adulthood
Glycogen storage disorders (GSD)
GSD type 2: Pompe’s disease
GSD type 3: debrancher deficiency
GSD type 4: Andersen’s disease
GSD type 5: McArdle’s disease
GSD type 7: Tarui’s disease
GSD type 9: phosphorylase kinase deficiency
Fatty acid oxidation disorders
Carnitine palmitoyltransferase II deficiency
Very-long-chain acyl-Coenzyme A dehydrogenase deficiency
Multiple acyl-Coenzyme A dehydrogenase deficiency
Disorders of purine metabolism
Myoadenylate deaminase deficiency
Mitochondrial disorders
In this review, we will focus on conditions involving defective glycogen metabolism (glycogenosis or glycogen storage disorders (GSD)) or fatty acid metabolism (lipidoses, fatty acid oxidation disorders). This is because (a) they may present in adulthood, usually to neurologists, but also to acute physicians and rheumatologists and (b) in adults they usually lack the phenotypic multisystem clues that accompany the childhood onset forms of metabolic disease, or mitochondrial disease.
Metabolic myopathies are rare. Most conditions discussed here have orphan status, including McArdle’s disease (ORPHA:368), carnitine palmitoyltransferase II (CPTII) deficiency (ORPHA:228302) and late-onset Pompe’s disease (ORPHA:420429). However, collectively they are relatively common in neurological practice, and a frequent cause of diagnostic concern and confusion. McArdle’s disease (GSD type 5) and Pompe’s disease (GSD type 2) each affect around 1 in 40 000 people.1 2 However, these prevalence estimates are the subject of much debate, particularly regarding the relevance of apparently pathogenic mutations in asymptomatic or pauci-symptomatic individuals.3 In addition, recent advances in molecular genetic diagnostic techniques and increased screening of ‘at-risk’ individuals have identified increasing numbers of patients.4 5
There are now specific treatments available for certain metabolic myopathies (eg, Pompe’s disease). It is therefore vital to be aware of their key clinical features to ensure relevant patients are identified early, accurately diagnosed and offered appropriate treatment. Salford (Manchester, UK) is the largest single-site centre for adult inherited metabolic disorders in the UK. From our experience, we present our insights into some of these disorders and address other conditions that can present in a ‘pseudometabolic’ fashion.
When to suspect a metabolic myopathy
When approaching a patient with a suspected metabolic myopathy, the overall aim is to:
Separate those that might have a ‘genuine’ metabolic myopathy, from those with other conditions with a ‘pseudometabolic’ mode of presentation (which may mimic a metabolic myopathy).
Determine clinically the most likely affected biochemical process (eg, glycogen storage or fatty acid metabolism).
Identify which patients should be investigated further and by what methods (eg, muscle biopsy, genetics, etc).
Identify those patients with conditions for which there are pharmacological and/or other treatments available (eg, Pompe’s disease).
Offer genetic counselling to relatives who may also be affected.
Patients with a metabolic myopathy often report episodes of exercise intolerance, fatigue, muscle pain and weakness, but are often free of symptoms at the time of their review in the outpatient clinic. The clinician needs some detective skills to identify clues to the underlying diagnosis. For example, while the current serum creatine kinase might be normal, identifying previous fluctuations in serum creatine kinase by going through old biochemistry reports might be critical in making a diagnosis. Reviewing, with consent, the medical records of relatives can also provide vital clues.
As with any other diagnostic evaluation in neurology, the initial focus is on the history and clinical examination (box 2).
Key points in history and examination findings in the metabolic myopathies
History
Episode(s) of rhabdomyolysis
Episodes(s) of pigmenturia
Exercise intolerance and fatigue
Symptoms arising after periods of fasting (eg, for religious reasons)
Prominent myalgia, cramps and stiffness
Consanguinity and affected family members
Examination
Contractures (fixed or transient; see below)
Ptosis or ophthalmoplegia (mitochondrial disorders)
Clues to alternative diagnosis (eg, calf pseudohypertrophy seen in Becker’s muscular dystrophy)
Important aspects of the history
Exercise intolerance and myalgia
Exercise intolerance due to fatigue, muscle pain (myalgia) and cramping are key metabolic symptoms. However, the relevance of such symptoms is often overlooked. For example, ‘the sporting stoic’ may have suffered from long-standing exercise-induced myalgia with a ‘second wind’ phenomenon, but assumed this to be normal.6 They might seek medical advice only after having had an episode of severe rhabdomyolysis or painful ischaemic contracture. Myalgia, or even rhabdomyolysis, experienced in the context of intense unaccustomed exercise is usually benign. However, ongoing or recurrent symptoms in a single team member, in contrast to team-mates who have developed tolerance to the level of exertion, may provide a clue to unmask an underlying metabolic defect.7
Once the patient has disclosed a history of exercise-induced myalgia, it is important to establish the type of activity that produces symptoms.
Symptoms precipitated by short bursts of high-intensity exertion, usually within around 5 min, should lead clinicians first to consider glycogen storage disorders. A ‘second wind’ phenomenon may then occur in McArdle’s disease, with a resolution of pain and a regaining of muscle strength after resting for around 10 min, at which point further exertion can take place without symptom recurrence.
Symptoms precipitated by prolonged periods of low-intensity (‘endurance’) exercise are more typical of the fatty acid oxidation disorders, but can also occur in mitochondrial myopathies.8
Rhabdomyolysis
Patients may experience pigmenturia without recognising its significance ("I thought I was just dehydrated"). Additionally, a false-positive urine dipstick for blood in such cases can lead to misdiagnosis as a urinary tract infection or other urological problem. In such cases, myoglobin (extruded from damaged skeletal muscle) cross-reacts with the haemoglobin assay on the dipstick.
Rhabdomyolysis has numerous causes, including infections (particularly influenza), seizures, trauma, drug toxicity and diabetic ketoacidosis.9 other symptoms of metabolic dysfunction, such as headache, fever, vomiting and malaise may make it more difficult to identify the underlying cause. We have diagnosed a metabolic myopathy in several patients first thought to have viral myositis because of these accompanying symptoms (box 3).
Mitochondrial cytopathy presenting as rhabdomyolysis
A man aged 62 years presented to hospital with a swollen painful leg, dark urine and a low-grade fever. His serum creatine kinase was markedly raised at 24 000 U/L (normal 24–170). There was no clear provoking factor for this presentation. He was managed with intravenous fluids and was discharged home with a diagnosis of ‘viral myositis’.
On follow-up in neurology clinic, we noted he also had slowly worsening bilateral ptosis, complex ophthalmoplegia and sensorineural deafness. He also had been diagnosed with diabetes mellitus a few years previously.
Muscle biopsy identified excess cytochrome oxidase-negative fibres, suggesting a mitochondrial disorder. Long-range PCR showed evidence of mitochondrial rearrangements, although formal DNA analysis was negative. We trialled him on ubiquinone without significant effect but he remains stable on repeated review.
Rhabdomyolysis in people with a history of preceding unaccustomed exercise (‘too much, too fast, too soon’) is usually a physiological exercise-induced rhabdomyolysis.7 A famous example of this involved the McMinnville (Oregon, USA) high-school football team in August 2010. Here, 43 footballers were put through a vigorous preseason training regimen (‘immersion camp’); 12 were admitted to hospital with rhabdomyolysis and 3 required fasciotomies for triceps compartment syndrome.10 It is obviously unlikely that almost 30% of these footballers had a genetic predisposition for the development of rhabdomyolysis, and in our experience, genetic testing in this setting has a low yield for identifying any underlying disorders. However, recurrent episodes, persistent symptoms and a positive family history all increase the likelihood of identifying an underlying rhabdomyolysis predisposition.7 Other triggers of rhabdomyolysis in susceptible people include fasting and intercurrent illness, particularly in those with defects of fatty acid oxidation.11
Family history
Simply asking ‘does anything run in the family?’ may not be adequate. We recommend drawing a family tree and directly questioning about the possibility of consanguinity. This is of special importance given that many of the disorders considered here have an autosomal recessive inheritance pattern. Patients from populations where genetic diversity is limited are also at higher risk. Until recently, many metabolic muscle diseases were not well documented or were difficult to confirm. Affected siblings with metabolic disorders may also show variations in clinical presentation. Questions regarding family members should therefore not be limited to individuals diagnosed, and to those with any suggestive symptoms, including exercise intolerance and early use of walking aids.
Developmental milestones
Although often presenting in adulthood, many patients with a metabolic myopathy have had long-standing symptoms dismissed by the patient, their parents and physicians alike. ‘Growing pains’ is a particularly common misdiagnosis.12 Several genetic myopathies may present in a ‘pseudometabolic’ fashion (box 4). It is therefore important to question the patient (and relatives) carefully about other symptoms of neuromuscular disease, such as delays in walking ability or poor performance at school sporting events.
Other disorders that may present in a ‘pseudometabolic’ fashion in adults
Becker’s muscular dystrophy (including manifesting carriers)
Limb girdle muscular dystrophies, eg, FKRP myopathy (limb girdle muscular dystrophy type 2I)
‘Congenital’ myopathies, eg, RYR myopathy (central core disease)
Myotonic myopathies: proximal myotonic myopathy (myotonic dystrophy type 2)
Vitamin D deficiency, thyroid dysfunction
Examination findings
Neurological examination in people with a metabolic myopathy is often normal, particularly between episodes of symptoms. However, a thorough examination is essential as this can sometimes identify subtle diagnostic clues. People with McArdle’s disease (GSD type 5) may develop permanent weakness; up to 40% of older patients with this condition have proximal weakness.12 Pompe’s disease (GSD type 2) is characterised by fixed weakness of axial musculature (particularly paraspinous muscles) and proximal leg muscles.2 Scapular winging and ventilatory muscle weakness can also occur (box 5). Fixed weakness is less common in the fatty acid oxidation disorders.
Scapular winging in late-onset Pompe’s disease (GSD type 2)
A man aged 49 years attended the neurology clinic with a 14-year history of hip and lower back pains. His friends had commented that he had an unusual posture. There was no family history of neuromuscular disorders.
On examination, he had marked scapular winging and atrophy of his paraspinal muscles. There was marked proximal limb weakness. His posture was characterised by an exaggerated lumbar lordosis. His quadriceps were also bilaterally wasted. His serum creatine kinase was raised at 312 U/L (normal 24–170) and an electromyography showed subtle myopathic changes. The provisional diagnosis was of a limb girdle muscular dystrophy.
He was referred to the neuromuscular clinic and further exploration of the history revealed that he had struggled with sports from a young age, suggesting at a slowly progressive myopathic disorder. Enzymology and later formal genetics confirmed a diagnosis of late-onset Pompe’s disease.
We started him on enzyme replacement therapy and he continues to be mobile on recent reviews.
‘Pseudometabolic’ syndromes
Other genetic myopathies that can mimic a metabolic myopathy may be associated with fixed weakness (box 4). For example, Becker’s muscular dystrophy, increasingly recognised in a mild adult-onset form, can present with calf pain on exertion and proximal arm and leg weakness. Manifesting female carriers of Becker’s muscular dystrophy can also create diagnostic confusion, and calf cramping may be the only symptom.13
Myotonic dystrophy type 2 (or proximal myotonic myopathy (PROMM)) can also present with exercise-induced myalgia, cramping, stiffness and fatigue (box 6).14 The onset is typically in the third decade. Overall, myotonic dystrophy type 2 tends to have a milder phenotype type 1, particularly in regard to cognitive impairment, and thus the patient may evade accurate diagnosis for some time. Proximal limb weakness, and sometimes facial muscle weakness, cataracts, deafness and heart rhythm abnormalities are important clinical clues to the diagnosis. The electromyogram (EMG) may show myotonic discharges, although these can be easily missed and may also occur in Pompe’s disease.
Myotonic dystrophy type 2 presenting as a mimic of metabolic myopathy
A woman aged 50 years attended the neuromuscular clinic with suspected late-onset myopathy. She had no relevant personal or family history, and had been developmentally normal as a child. She described an 8-year history of progressive proximal lower limb weakness, and a 6-month history of myalgia in her thighs.
On examination, she struggled to stand from a squatting position with her arms folded, and there was iliopsoas weakness (Medical Research Council grade 4+/5) with normal distal lower limb strength. Her electromyogram (EMG) showed myopathic changes in the iliopsoas and quadriceps muscles. Her serum creatine kinase was only mildly elevated at 198 U/L (normal 24–170) and Pompe’s disease enzymology was negative. MR imaging of her lower limb muscles showed slight fatty atrophy within the gluteus maximus bilaterally but no other significant abnormality. A muscle biopsy showed a both neurogenic and myopathic abnormalities, with no degenerative or dystrophic features.
A repeat EMG 1 year later showed some myotonia in addition to myopathic features, raising the possibility of proximal myotonic dystrophy type 2. This was confirmed by genetic analysis of the CNBP (ZNF9) gene. She was referred to cardiology for screening for potential cardiac conduction defects, and underwent defibrillator insertion due to episodes of documented ventricular tachycardia on testing. Several family members were screened and one of her sisters was found to have the same mutation.
Several recessive limb girdle muscular dystrophies (LGMDs) can mimic a metabolic myopathy, including dysferlinopathies (LGMD2B), FKRP myopathy (LGMD2I), ANO5 myopathy (LGMD2L) and the sarcoglycanopathies (LGMD2C and LGMD2E).7 Myopathies associated with RYR1 gene mutations (central core disease) are usually classified with congenital myopathies, but have a highly heterogeneous phenotype, including a late-onset pseudometabolic presentation.15 Patients are at risk of malignant hyperthermia—a life-threatening combination of fever, rhabdomyolysis and rigidity occurring after exposure to certain medications, particularly inhaled anaesthetics. This may even occur without clinical evidence of myopathy.
There are several other catches. For example, thyroid dysfunction and vitamin D deficiency can present with symptoms such as cramping, exercise intolerance and a raised serum creatine kinase. Finally, we have encountered patients referred with ‘?myositis’ who ultimately have been diagnosed with metabolic myopathies, including McArdle’s disease and Pompe’s disease. In general, a diagnosis of ‘seronegative polymyositis’ should ring alarm bells (box 7, case 1).16
Two cases of late-onset Pompe’s disease presenting with respiratory failure
Case 1:
A man aged 25 years was referred to the neuromuscular service after presenting with type 2 respiratory failure. He had a 3-month history of increasing tiredness and drowsiness and had been found to be hypoxic with a marked hypercapnia, with an elevated serum creatine kinase.
He was initially thought to have polymyositis and treated with corticosteroids. His respiratory state improved with bilevel positive airways pressure ventilation. A quadriceps muscle biopsy showed glycogen accumulation with membrane-bound vacuoles, raising the possibility of Pompe’s disease.
On further questioning, he reported delayed motor milestones as a child and that he had struggled with sports. On examination, he had marked paraspinal and periscapular muscle wasting with hamstring, calf and anterior leg muscle wasting.
Genetic testing confirmed a diagnosis of late-onset Pompe’s disease and he started enzyme replacement therapy. He continues to use overnight continuous positive airways pressure at night to help with ventilation.
Case 2:
A man 50 years was referred to the neuromuscular clinic following investigations after a hypertensive intracranial brain haemorrhage. He had been slow to wean from non-invasive ventilation and he had continued to require continuous positive airways pressure overnight ever since. He also reported delayed motor milestones as a child and recalled having scapular winging. He struggled with sports at school.
On examination, he had generally reduced muscle bulk with paraspinal atrophy and bilateral scapular winging.
A quadriceps muscle biopsy showed features consistent with acid maltase deficiency and genetic testing confirmed late-onset Pompe’s disease. He was started on enzyme replacement therapy and continues to do well on recent review.
Contractures—fixed or transient?
It is important to distinguish between fixed and transient muscle contractures; both may occur in muscle diseases.
Transient muscle contractures
These contractures are often painful and occur due to a failure to meet the demands of the energy-dependent process of muscle relaxation. Impaired calcium binding by the sarcoplasmic reticulum probably plays a role in this. These transient contractures classically occur in McArdle’s disease (GSD type 5) in the context of high-intensity anaerobic exercise. Prolonged contracture can cause muscle ischaemia and necrosis, and some patients may develop compartment syndrome necessitating urgent fasciotomy. Myoglobulinuria may result from muscle breakdown.
Fixed, painless muscle contractures
These contractures occur more commonly in some muscular dystrophies (eg, Becker’s muscular dystrophy). These probably result from a decrease in sarcomere numbers, which shortens the muscle length and increases their resistance to passive stretch. Fixed contractures may develop particularly in Pompe’s disease (GSD type 2), but can also follow episodes of rhabdomyolysis from any cause.
Ptosis, ophthalmoplegia and multisystem disease
Cardiac involvement is common in early onset Pompe’s disease (GSD type 2), but is less common in the late-onset form.17 Otherwise, cardiac disease generally does not occur in either the glycogen storage diseases or fatty acid oxidation disorders. The presence of heart involvement may be a clue to a metabolic myopathy mimic, such as Becker’s muscular dystrophy or myotonic dystrophy type 2. Ventilatory muscle weakness is common in late-onset Pompe’s disease and may present insidiously with nocturnal hypoventilation and type 2 respiratory failure (box 7).17
Identification of ptosis and ophthalmoplegia, which may be subtle and pass unrecognised for many years, may suggest a diagnosis of a mitochondrial disease. In addition to exercise intolerance, people with mitochondrial diseases often have hearing loss, epilepsy, cognitive impairment, neuropathy or retinopathy.
Investigations
Early involvement of an experienced multidisciplinary team—comprising neuromuscular specialists including neurologists and rheumatologists as well as neurophysiologists, metabolic medicine specialists and neuropathologists—is key to tailoring investigations, reducing costs and avoiding invasive investigations, where possible.
The serum creatine kinase should be checked. It is rarely normal in people with McArdle’s disease, even between episodes of symptoms. When considering ‘pseudometabolic’ conditions, the serum creatine kinase provides a rough guide to the differential diagnosis.18 However, the result must be interpreted in the context of any recent exercise and medications that might cause elevations (eg, statins). In our experience, a serum creatine kinase persistently above 5000 IU/L is unlikely to reflect a metabolic disorder.
EMG and muscle biopsies can yield important clues to help support a diagnosis of a metabolic myopathy, or to highlight clues to an alternative diagnosis (eg, myotonic discharges on EMG that might suggest myotonic dystrophy, or dystrophic change on muscle biopsy). There is an increasing shift towards genetic testing as the diagnostic investigation of choice in those with a suspected metabolic myopathy. This is already commonplace for McArdle’s disease (GSD type 5), where the history often strongly suggests this particular disorder. Furthermore, next-generation ‘gene panels’ allow for rapid and economically viable screening of multiple genes associated with metabolic myopathy, reducing the need for repeated candidate gene testing. It can be highly beneficial to use such gene panels, but sometimes they produce results that are difficult to interpret (box 8).
Carnitine palmitoyltransferase II (CPTII) deficiency presenting with rhabdomyolysis
A woman aged 26 years was referred to the neuromuscular clinic after an episode of rhabdomyolysis triggered by exercise, with an initial serum creatine kinase of 59 000 U/L. In retrospect, she had been having exercise-induced myalgia and myoglobulinuria for a few years despite being very fit and active. Her myalgia tended to happen 40–120 min into exercise and then would persist for days afterwards. There was no ‘second wind’ phenomenon. Examination was normal and a free carnitine and acylcarnitine profile was abnormal.
We did further tests in the form of culture skin fibroblast analysis, which showed features suggesting CPTII deficiency. Genetic analysis using a next-generation sequencing panel, showed that she was homozygous for CPTII gene mutations, but interestingly she also carried a very-long-chain acyl-CoA dehydrogenase deficiency gene mutation (of undetermined significance).
She has remained well on follow-up and has been managed with a dietary regimen of frequent carbohydrate meals and medium-chain triglyceride oil supplementation before exercise.
An overview of some diseases to consider in adults
Glycogen storage disorders
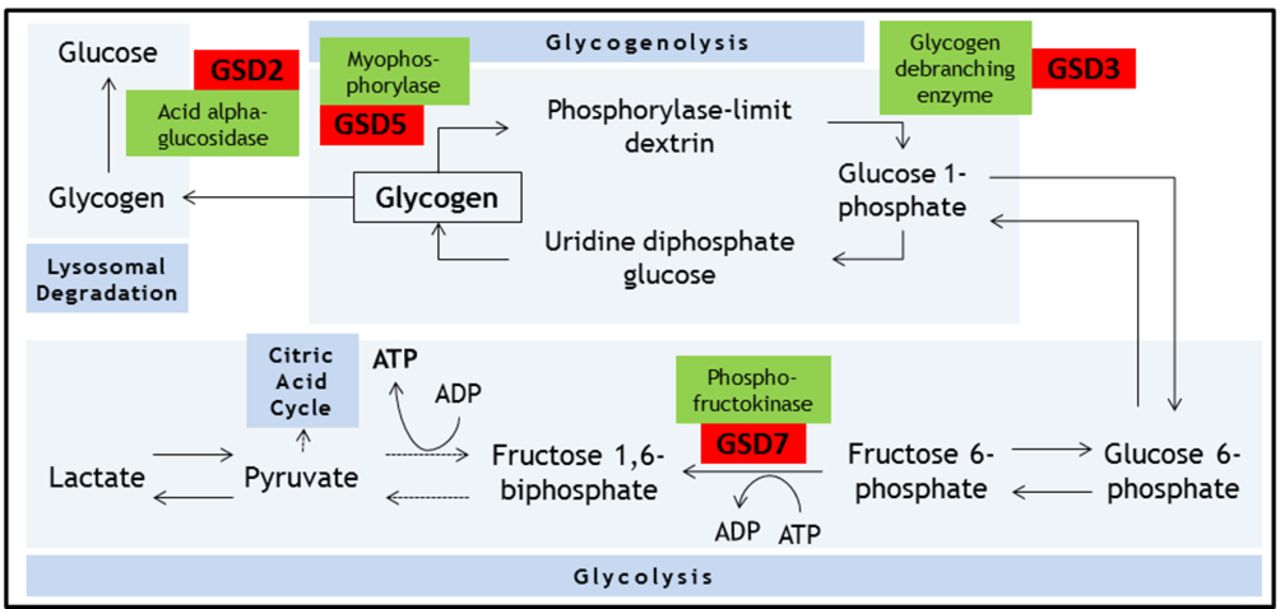
Energy for skeletal muscle contraction is generated via several complex and interlinked metabolic pathways, involving carbohydrates, fats and proteins. During isometric muscle contraction, energy for myofibre contraction depends on anaerobic glycolysis (breakdown of glucose to produce ATP).19 Raw materials for this process rely on mobilising intracellular glycogen stores (glycogenolysis), as free diffusion of glucose across the sarcolemma is not possible (due to the increased pressure within this compartment during isometric contraction). There are reports of disorders associated with defects in most enzymatic steps in these metabolic processes (figure 1).

Summary of the biochemical processes of glycogenolysis, glycolysis and lysosomal degradation. Key glycogen storage disorders that can present in adulthood and associated enzymes are shown. Dotted arrows represent omitted intermediate steps. GSD, glycogen storage disorder.
McArdle’s disease (GSD type 5)
Background
Dr Brian McArdle described this condition in 1951; it is the most common disorder of carbohydrate metabolism.20 McArdle’s disease results from mutations in both alleles of the PYGM gene. Inheritance is in an autosomal recessive pattern and males and females are affected with equal prevalence. PYGM encodes the skeletal muscle specific isoform of myophosphorylase (cardiac and hepatic isoforms remain unaffected).
Myophosphorylase catalyses the conversion of glycogen to glucose 1-phosphate (glycogenolysis) (figure 1). Without myophosphorylase, the glucose-hungry process of glycolysis is starved of a key source of its substrate. As a result, energy production depends on either direct influx of glucose from the bloodstream (ie, bypassing the defective step), or on other metabolic process (eg, fatty acid metabolism).
Clinical features
Symptoms of McArdle’s disease usually develop in childhood, although the presentation may not be until early adulthood. Presentation in late adulthood is also well recognised. All patients report exercise intolerance and many report postexertional pigmenturia or previous bouts of rhabdomyolysis. Most patients report experiencing the ‘second wind’ phenomenon. McArdle’s disease is often misdiagnosed at first presentation: a recent retrospective study in London showed 45 of their 50 patient cohort received an erroneous first diagnosis with a median delay to diagnosis of 29 years (range 0–68).21
McArdle’s disease shares clinical features with other glycogen-storage disorders, especially Tarui’s disease (GSD type 7), which has infantile and late-onset forms. Tarui’s disease is associated with haemolytic anaemia due to involvement of phosphofructokinase isoforms present in reticulocytes, and thus the reticulocyte count and serum bilirubin are often raised. Another important difference is the differing response to administering glucose before exercise. People with McArdle’s disease’s benefit (in terms of exercise tolerance) from this practice, as their metabolic defect is upstream; in effect, the defective step is bypassed. In contrast, people with Tarui’s disease do not benefit as they cannot use glucose efficiently; in fact, the increased plasma glucose concentration decreases the availability of other sources of fuel (fatty acids and ketones), actually making the situation worse.19 This is described as the ‘out of wind’ phenomenon.22 23
Investigations
The serum creatine kinase is usually raised, even after prolonged periods of rest. In the past, the forearm ischaemic lactate test (as originally described by McArdle) was used to confirm the diagnosis. A failure of exercise-associated lactate production in the face of normal or exaggerated ammonia production is the expected finding in McArdle’s disease. However, such exertion can induce painful transient muscle contractures and produce false-negative results (eg, because of poor effort) or false-positive results (eg, the diagnosis is in fact another enzyme defect that produces the same outcome). Thus, this test is now rarely performed. However, supervised exercise testing to observe the ‘second wind’ phenomenon can be useful and a supervised cycle test may give highly sensitive and specific results.24 Supervised exercise testing can also help with disease monitoring.25
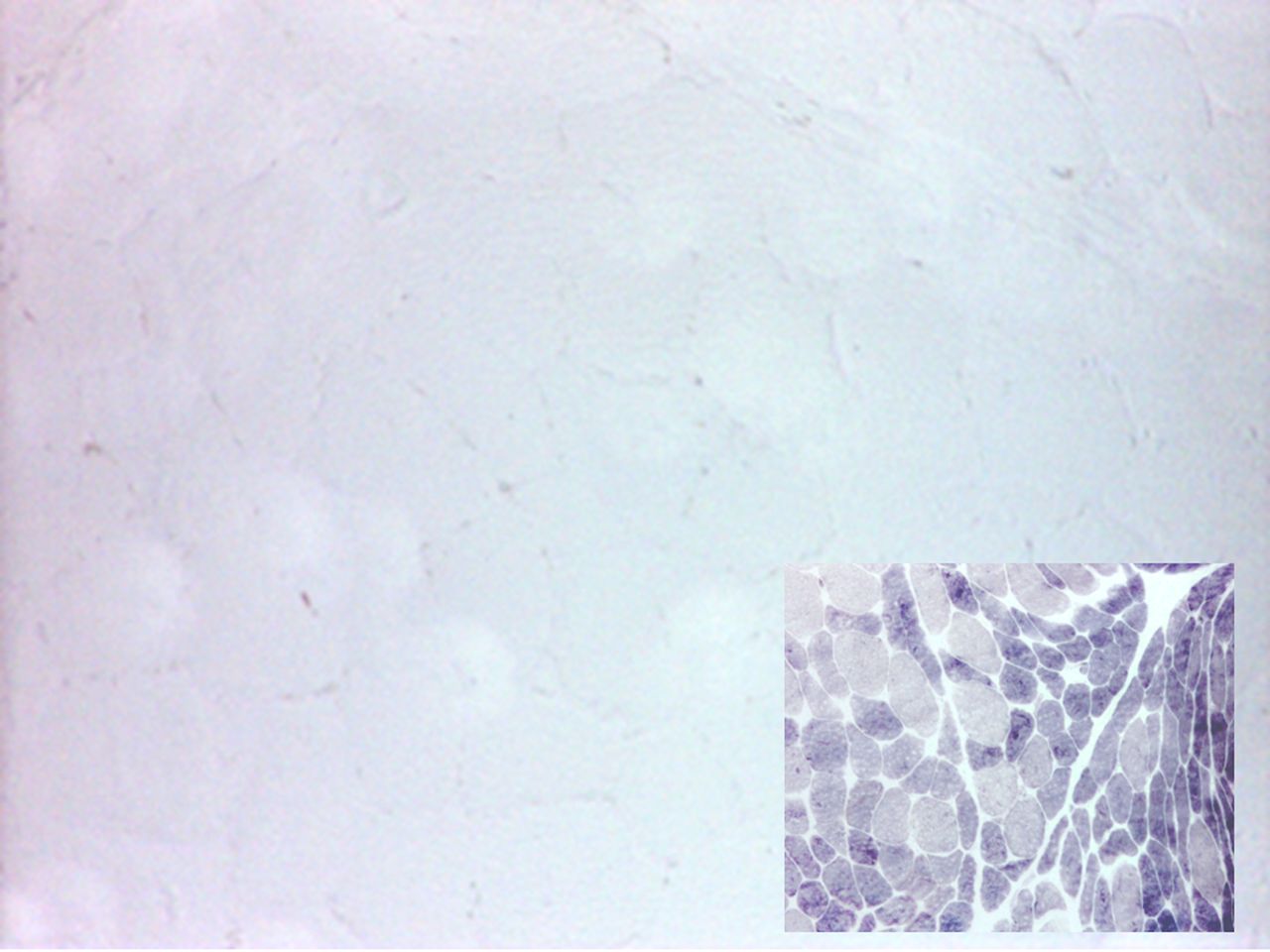
Muscle biopsy is also now less regularly performed, particularly in those with a typical history, but shows increased periodic acid-Schiff (PAS) staining (reflecting increased concentration of intramuscular glycogen that cannot be catabolised) and an absence of myophosphorylase staining (figure 2). Quantitative enzyme activity testing shows very low or undetectable residual activity.
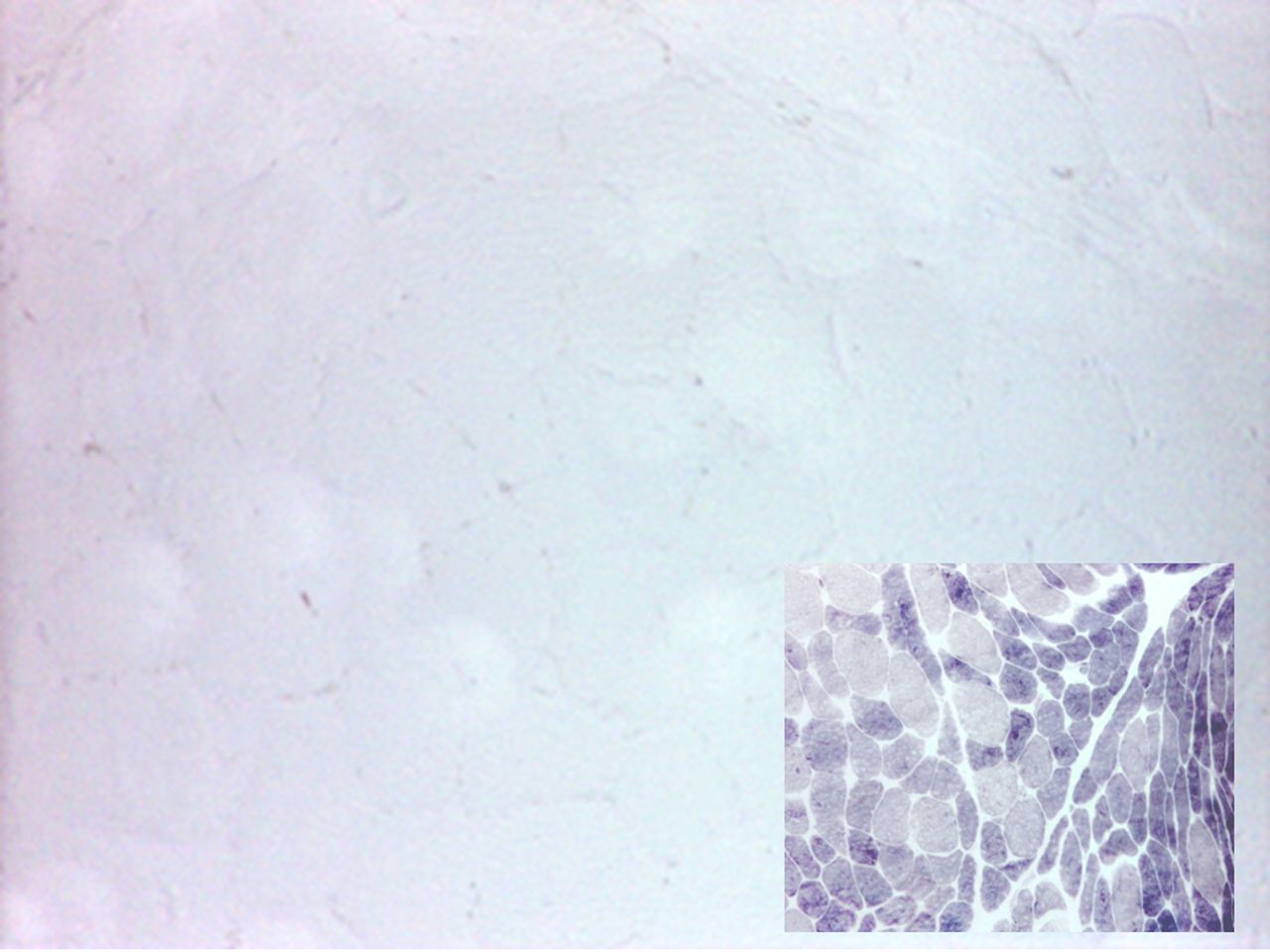
Quadriceps muscle biopsy from a patient with McArdle’s disease; myophosphorylase staining is absent; control muscle (inset) shows normal staining; type 2 fibres are more intensely stained.
Many now use genetic testing in the first instance in people with typical clinical features of McArdle’s disease. The usual first step is ‘hot spot’ analysis, which identifies most common mutations, although practice varies between centres. The p.Arg50Ter (sometimes termed R50X) mutation occurs in up to 85% of Caucasian probands and the p.Gly205Ser in up to a further 10%.26 If these are negative, then patients may require sequence analysis and deletion/duplication analysis; new mutations are regularly being described.27 28 We have encountered several patients with typical features of McArdle’s disease who are heterozygous for mutations in PYGM. At least in some cases, it seems there is either an additional inconspicuous mutation in the apparently unaffected allele which has evaded detection, or there are mutations in alternative genes involving related metabolic pathways (digenic inheritance, or ‘double trouble’).19
Management
As with other rare diseases, McArdle’s disease has a frustratingly limited therapeutic evidence base, and progress has been slow. A Cochrane review from 2014 found no new trials since the previous review in 2004.29 The management currently relies on attempting to reduce the risk of developing episodes of rhabdomyolysis. This can be partially achieved by dietary methods (a mixture of ‘background’ complex carbohydrates together with ‘simple’ carbohydrates before exertion) and by supervised aerobic exercise programmes that help to condition muscles for fatty acid metabolism.30 Patients should be advised to avoid anaerobic activity patterns, particularly those involving frequent eccentric (lengthening) or isometric muscle contractions (eg, squats, lunges, weight lifting). It is best to involve a specialist metabolic multidisciplinary team, including dieticians and physiotherapists.
Late-onset Pompe’s disease (GSD type 2)
Background
Pompe’s disease (also known as GSD type 2 and ‘acid maltase deficiency’) is also autosomal recessive and results from mutations in the GAA gene, which codes for lysosomal acid alpha-glucosidase (acid maltase) (figure 1). Defects in acid maltase function lead to a toxic build-up of intralysosomal glycogen, initiating a cascade of events that culminates in cell death. There are both early and late-onset forms, approximately correlating with enzyme activity levels.2 In the most severe congenital cases of Pompe’s disease there is practically no measurable enzyme activity, while adults usually have around 20% residual enzyme activity. Recently, clinicians have had a lower threshold for screening patients, particularly those with cryptogenic muscle disorders, using dry blood spot enzymology. In one series of adult patients with hyperCKaemia and/or limb girdle weakness, 2.4% were found to have Pompe’s disease.4
Clinical features
The most common symptom of late-onset Pompe’s disease is proximal muscle weakness, particularly in the lower limbs.31 Ptosis, paraspinal wasting and scapular winging are also common (figure 3). Patients may report exercise intolerance, but largely because of shortness of breath and fatigue, rather than due to the myalgia characteristic of McArdle’s disease. We have diagnosed Pompe’s disease in patients presenting with unexplained type 2 respiratory failure (sometimes in extremis). Another increasingly recognised complication of Pompe’s disease is dilative vasculopathy32 (figure 4). We therefore have a low threshold for screening for Pompe’s disease in patients with otherwise unexplained subarachnoid haemorrhage.

Asymmetrical scapular winging and paraspinal muscle wasting in a patient with Pompe’s disease.

Three-dimensional reconstruction of MR angiogram, showing marked dolichoectasia of vessels of the vertebrobasilar system and both internal carotid arteries.
Investigations
The serum creatine kinase may be raised up to 10 times the upper limit of normal, but may be normal.18 We now use dry blood spot enzymology as first line in patients with suspected Pompe’s disease to estimate residual acid maltase enzyme activity, since its sensitivity is higher than muscle biopsy.33 The heterogenous phenotype of Pompe’s disease means that many patients will have had a muscle biopsy to look for dystrophic change. In Pompe’s disease, the muscle biopsy may show a PAS-positive vacuolar myopathy (figure 5), but may be normal or show only non-specific abnormalities. Current practice generally is to confirm Pompe’s disease with molecular genetic testing of the GAA gene. This gives the ability to detect carriers of the disease (and so to offer them appropriate genetic counselling) and reduces the risk of misdiagnosis in those with acid maltase ‘pseudodeficiency’, which is especially common in Asian populations.31
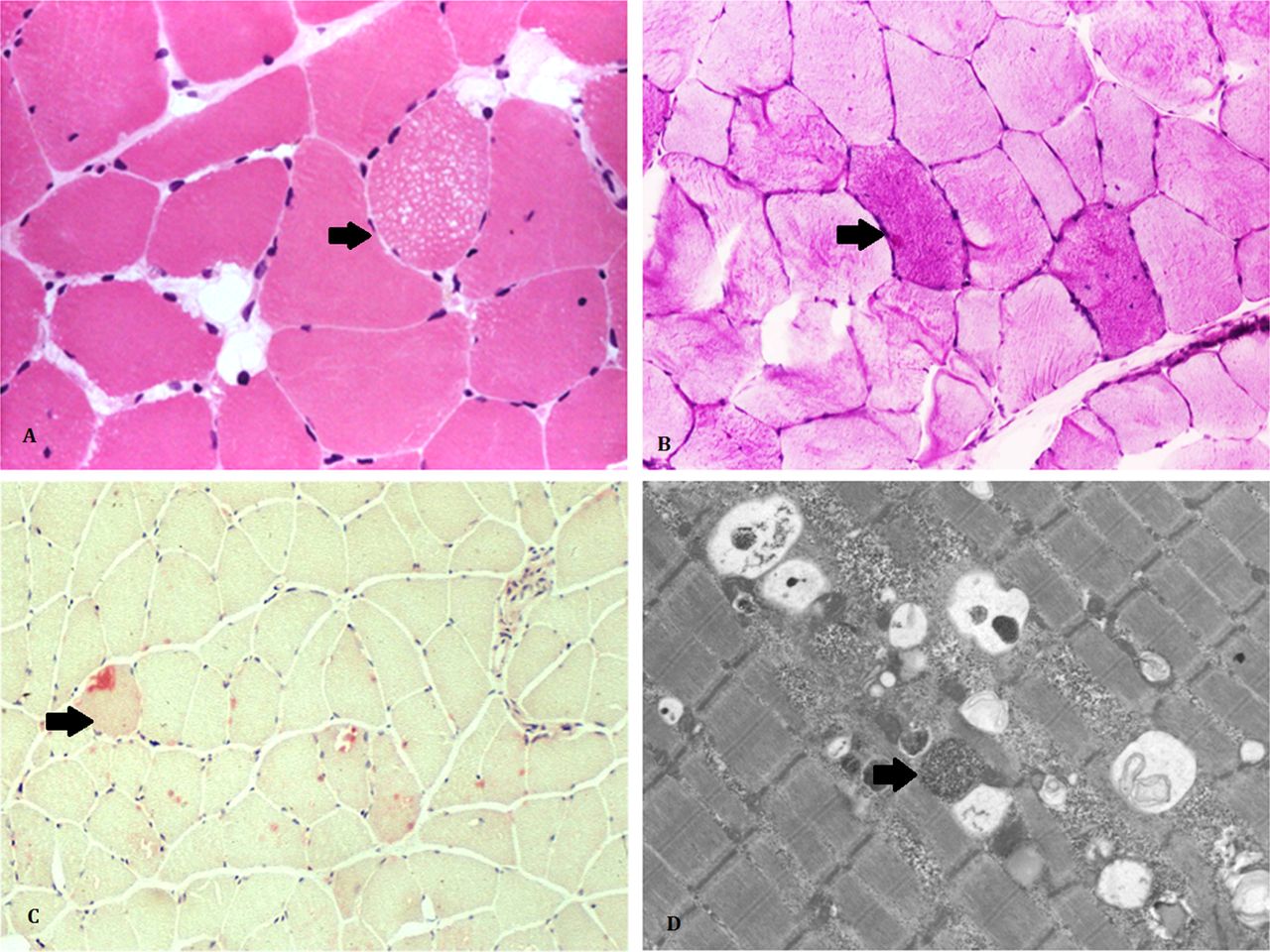
The muscle biopsy of people with late-onset Pompe’s disease often shows subtle changes including small sarcoplasmic vacuoles in a few fibres (arrow) (A, H&E x20) and slight increase in glycogen store (arrow) (B, periodic acid-Schiff x20); the special histochemical stain for acid phosphatase (arrow) (C, x20) highlights increase of lysosomes in a few fibres; ultrastructural examination can help to identify lysosome-bound glycogen (arrow) (D, Electron Microscopy x5000).
Management
There have been several clinical trials of enzyme replacement therapy for Pompe’s disease. Small open-label studies in childhood forms of the disease have shown benefit, including an improved chance of ventilator-free survival in treated patients.34 The true efficacy in adults with milder and more slowly progressive phenotypes remains the subject of debate. A randomised controlled trial of enzyme replacement therapy in adults, completed in 2010, showed improved performance on the 6 min walk distance test and stabilised pulmonary function in treated patients with late-onset disease.35 While enzyme replacement therapy clearly has some effect in adults over placebo, the question remains unanswered as to whether this translates into reduced morbidity (eg, requirement for ventilatory support or a wheelchair) and mortality.36 Enzyme replacement therapy is currently offered to all patients in the UK and there is a developing consensus regarding relevant start and stop criteria.37 A 5-year observational study of 22 adult-onset people with Pompe’s disease on enzyme replacement therapy showed that most sustained stable pulmonary function and mobility after 5 years of treatment.38
From a pragmatic perspective, physical therapy is important and patients require regular screening for respiratory failure, with involvement of ventilatory support teams where appropriate.
Fatty acid oxidation defects
Long-chain fatty acids, the major fraction of fatty acids in circulation, represent a key energy source for muscle during periods of endurance (prolonged, low intensity) exercise or in situations where the body makes efforts to spare glucose (and thus switch to fatty acid metabolism), such as during fasting.39 However, unlike short-chain and medium-chain fatty acids (which can freely diffuse across membranes), long-chain fatty acids require specific transporter proteins to traverse the outer and inner mitochondrial membrane before they can be catabolised (figure 6).
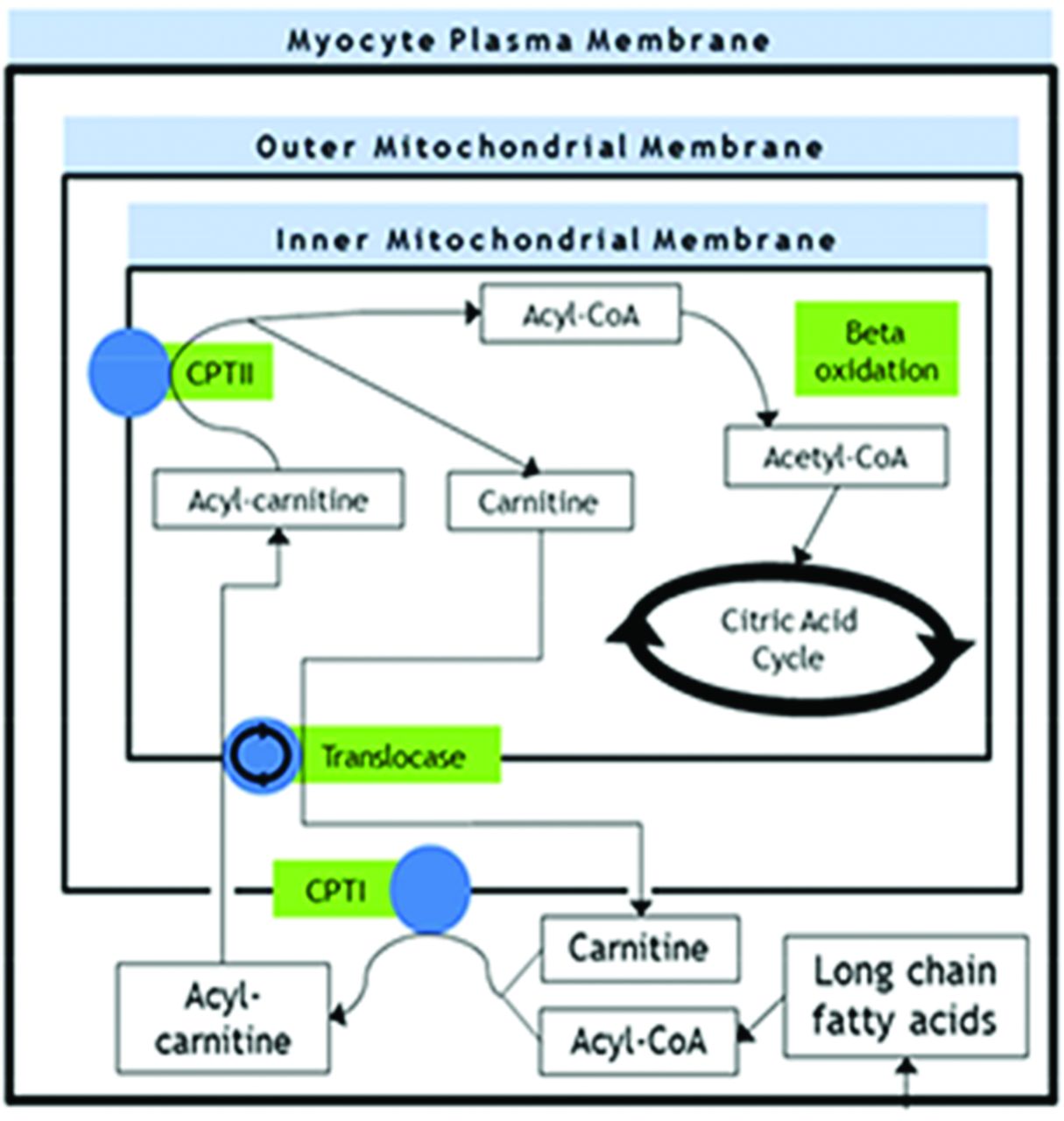
Summary of selected biochemical processes involved in fatty acid transport and oxidation. CPT, carnitine palmitoyltransferase.
Carnitine palmitoyltransferase II deficiency
Background
CPTII deficiency, caused by mutations in the CPT2 gene, is the most common fatty acid oxidation defect encountered in adults.39 CPTII is a membrane protein located on the inner mitochondrial membrane and is part of the mitochondrial long-chain fatty acid transportation system. This system shuttles long-chain fatty acids across the inner and outer mitochondrial membranes using carnitine recycling (figure 6). Ultimately, the system provides substrates for mitochondrial beta-oxidation and the citric acid cycle.
Clinical features
CPTII deficiency is autosomal recessively inherited, although with a male predominance, and may affect children and adults.39 Heterozygotes only rarely manifest the disease.40 The childhood forms of CPTII deficiency are multisystem disorders associated with liver failure and cardiomyopathy, whereas the late-onset form is usually restricted to skeletal muscle. Patients may complain of cramps and exercise intolerance before an episode of rhabdomyolysis prompts medical review.
Investigations
Investigations usually show an abnormal serum carnitine/acylcarnitine profile, with increased long-chain acylcarnitine (figure 7). ‘Ictal’ serum creatine kinase can be markedly elevated (up to 400 times the upper limit of normal) and can persist for some time after the inciting event. The ‘interictal’ (basal) serum creatine kinase is normal.6 Muscle biopsy is usually normal and CPTII deficiency does not cause accumulation of intramuscular lipid droplets.41 Muscle and other tissues, including cultured skin fibroblasts, show reduced CPTII enzyme activity. However, such assays are difficult to perform and there is no standardisation between centres. Thus, diagnostic practice is now shifting towards earlier use of genetic testing. This may start with targeted analysis of more common genetic defects (eg, p.Ser113Leu pathogenic variant found in 60% of those affected) before moving to more complex gene sequencing analysis.40

Acylcarnitine profiles for a patient with carnitine palmitoyltransferase II (CPTII) deficiency (above) compared with a normal subject (below). Note that the tracing in CPTII deficiency shows abnormal increases in the long-chain fatty acids (C10, C14:1, C14, C16:1, C16, C18:2, C18:1, C18) compared with the normal tracing, in keeping with the role of CPTII in the long-chain fatty acid metabolism pathway (see text and figure 6). With thanks to Alistair Horman and Teresa Wu (Willink Lab, Central Manchester Foundation Trust).
Management
Patients with CPTII deficiency require the involvement of specialists in metabolic diseases and an accompanying multidisciplinary team of dieticians and physiotherapists. Lifestyle modification to avoid periods of fasting and prolonged exercise is recommended. It probably helps to modify the diet with supplemented carbohydrates and medium-chain triglycerides, which are free to diffuse across the mitochondrial membrane without involving defective pathways.6 42 Despite in vitro studies suggesting that bezafibrate may help, a 2014 randomised controlled trial in people with CPTII deficiency and very-long-chain acyl-CoA dehydrogenase deficiency (VLCAD) found no benefit compared with placebo.43
Very-long-chain acyl-CoA dehydrogenase deficiency
Background
VLCAD catalyses the initial step of mitochondrial beta-oxidation of long-chain fatty acids with a chain length of 14– 20 carbons. Mutations in the ACADVL gene cause VLCAD deficiency.
Clinical features
Inheritance of VLCAD deficiency is autosomal recessive. The late-onset episodic myopathic form presents with intermittent rhabdomyolysis, muscle cramps, with or without pain and exercise intolerance.44 The adult-onset forms tend not to show the marked hypoglycaemia, cardiac or hepatic problems that occur in the early onset forms of the disorder.
Investigations
Comprehensive analysis of plasma or dried blood spot acylcarnitine during periods of metabolic stress (eg, fasting) using tandem mass spectrometry may be helpful. Common abnormal metabolites are C14:1, C14:2m C14 and C12:1. Genetic testing of the ACADVL gene may be performed and is increasingly being analysed as part of metabolic myopathy gene panels.
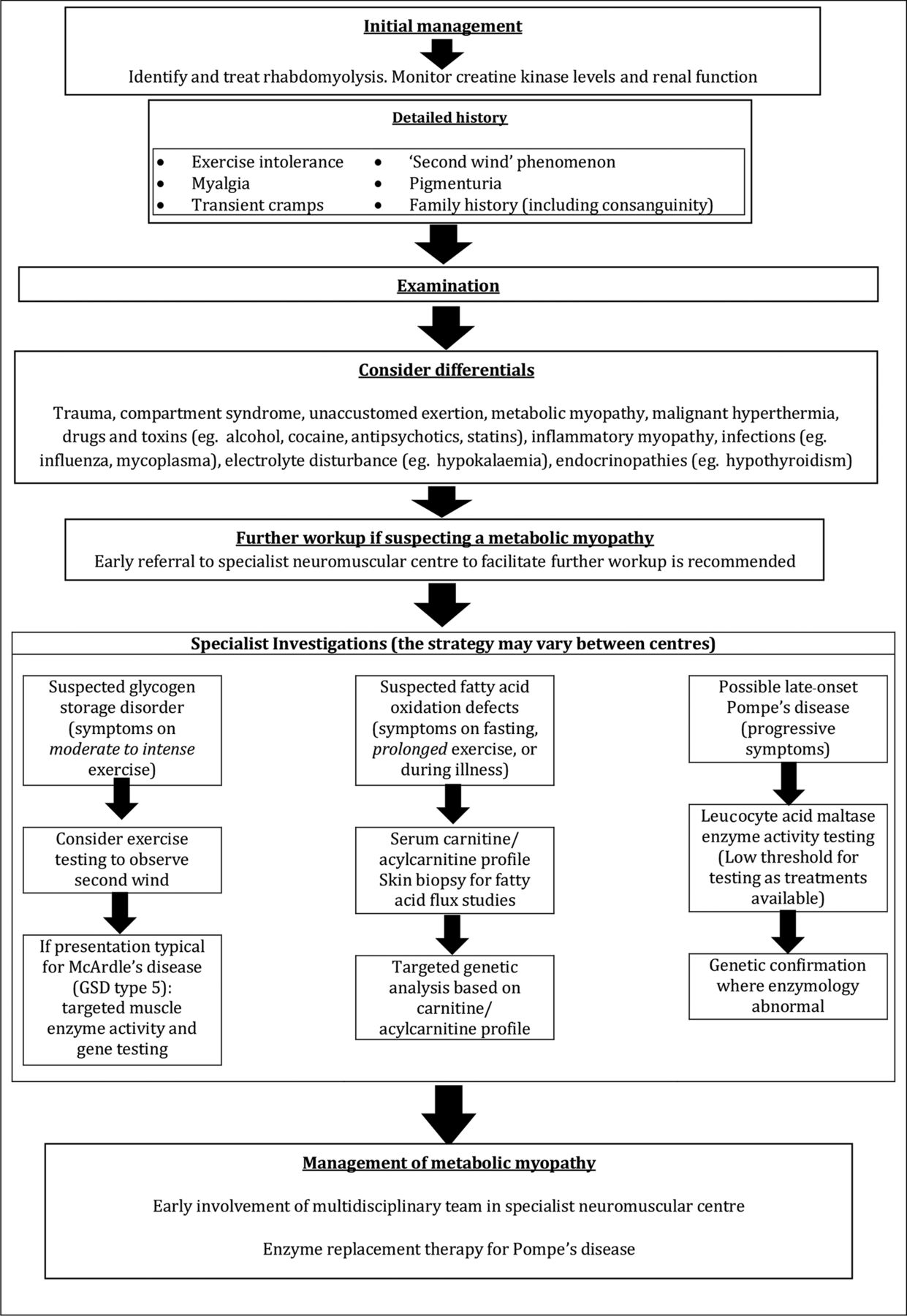
Summary of approach to a patient with suspected metabolic myopathy
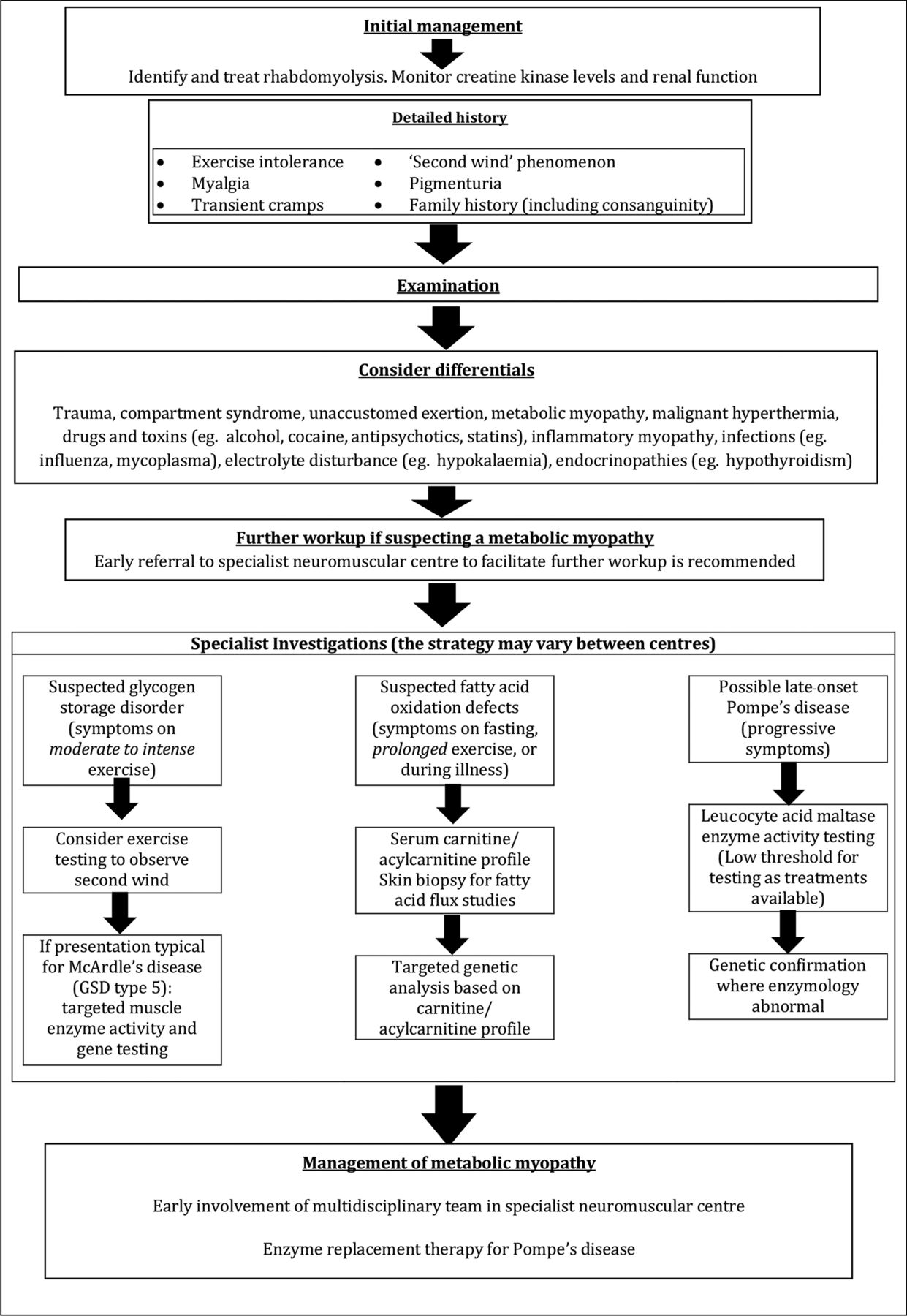
The diagnostic workup and subsequent management of patients with metabolic myopathies is complex and early referral to a specialist neuromuscular centre is strongly recommended. The sequence of investigative steps is strongly directed by the clinical history, particularly regarding the type of exercise that induces symptoms (high or low intensity), the presence (or otherwise) of a ‘second wind’ phenomenon and the presence (or otherwise) of fixed weakness or multisystem involvement.37
Figure 8 summarises a suggested clinical and therapeutic approach to such patients and table 1 reviews the key clinical and pathological features of some disorders discussed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of a suggested approach to patients presenting with rhabdomyolysis.
Key points
Metabolic myopathies are a diverse group of rare genetic disorders.
Muscle symptoms may be subtle and non-specific, and clinicians need a high index of suspicion for diagnosis, which is often delayed.
Standard laboratory tests may be normal or non-specific.
There are specific dietetic interventions, supplements and enzyme replacement therapies available for some conditions.
Main characteristics of a selection of glycogen storage disorders and fatty acid oxidation disorders
Acknowledgments
The authors thank Alistair Horman and Teresa Wu from the Willink lab, Central Manchester Foundation Trust for providing the acylcarnitine profile in Figure 7.
References
Footnotes
Contributors JBL created the initial draft. This was reviewed and updated by YSK. FR, RS and MER provided oversight and critical review of the manuscript. All authors approved the final version.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Jon Walters, Swansea, UK.
Other content recommended for you
- A diagnostic approach to recurrent myalgia and rhabdomyolysis in children
- Proximal muscle weakness
- Myeloperoxidase-positive ANCA-associated vasculitis presenting as myalgia, proximal weakness and a normal CK
- Emotionally-intense situations can result in rhabdomyolysis in McArdle disease
- Muscle disease
- Muscle biopsy: what and why and when?
- Muscle diseases: mimics and chameleons
- McArdle disease: a clinical review
- Muscle cramps and contractures: causes and treatment
- Genetic neuromuscular disease