Article Text

Abstract
Although sarcoidosis is rarely confined to the nervous system, any neurological features that do occur frequently happen early in the course of the disease. The most common neurological presentation is with cranial neuropathies, but seizures, chronic meningitis and the effects of mass lesions are also frequent. The diagnostic process should first confirm nervous system involvement and then provide supportive evidence for the underlying disease; in the absence of any positive tissue biopsy, the most useful diagnostic tests are gadolinium enhanced MRI of the brain and CSF analysis, although both are non-specific. The mainstay of treatment is corticosteroids, but these often have to be combined with other immunosuppressants such as methotrexate, hydroxychloroquine or cyclophosphamide. There is increasing evidence that infliximab is a safe treatment with good steroid sparing capacity.
Statistics from Altmetric.com
Sarcoidosis is a multisystem granulomatous disease usually diagnosed between the ages of 20 and 40 years. It has a propensity for the lungs, anterior uvea, lymph nodes and skin, but any organ can be affected resulting in an illness that may range from mild to life threatening. It has a prevalence of 40 per 100,000, although in certain racial groups such as West Africans it can be substantially higher.1,2 The cause remains speculative, but the documented ethnicity, histocompatibility associations and familial case reports suggest an underlying genetic predisposition.3,4 Also implicated in the pathogenesis are perhaps certain pathogens such as mycobacteria and propionobacterium acnes which may trigger the immunological effects—a dichotomy of heightened accumulation and activation of CD4+ helper T cells and macrophages in the affected organs (with release of various cytokines and proinflammatory factors) and depressed systemic cellular immunity.5,6 The pathological hallmark of the disease is the presence of multiple non-caseating epitheloid granulomas (structured masses of activated macrophages and their derivatives).
The Danish ophthalmologist, Heerfordt, first reported neurological manifestations of the illness in 1909 in his description of “uveoparotid fever” complicated by cranial neuropathies9 (although it has been suggested that long before this, sarcoidosis may have been responsible for the bewildering and complex medical condition that plagued Ludwig van Beethoven10–13). In practice, neurological involvement is a relatively rare though much feared complication. The current literature suggests that less than 10% of patients with sarcoidosis develop clinical evidence of neurological disease, with a course that may be acute, subacute or chronic with insidious onset.14–16 However, autopsy studies show a significant proportion with subclinical disease, and suggest that only 50% of cases are being diagnosed ante mortem.17,18 Although sarcoidosis is rarely confined to the nervous system, its neurological features frequently occur early in the course of the disease leading to diagnostic confusion.15,19
THE MANIFESTATIONS OF NEUROSARCOIDOSIS
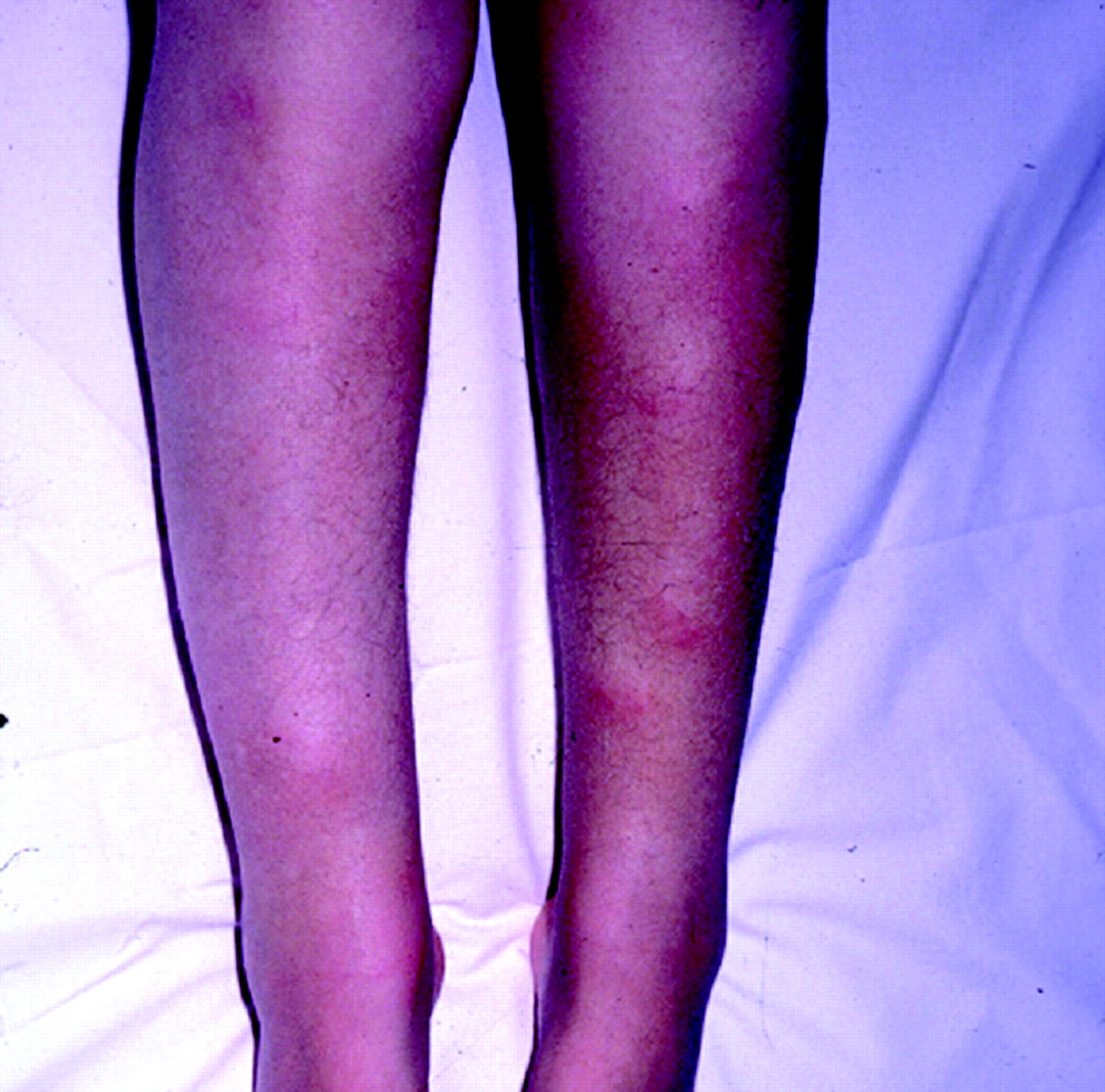
Our knowledge of the clinical features of neurosarcoidosis is derived from a handful of relatively large case series published over the last 40 years.14–22 Most have relied for diagnosis on the presence of recognisable neurological symptoms and signs in the context of a granulomatous multisystem disease, often supported by a tissue diagnosis from organs such as the liver, lungs (hilar nodes and/or parenchymal involvement) and skin (particularly erythema nodosum; fig 1), but only infrequently with nervous tissue confirmation.

Typical erythema nodosum in sarcoid.
Cranial neuropathies
This is the commonest manifestation seen in approximately 50–75% of patients with neurosarcoidosis.15,19,20 It may be caused by increased intracranial pressure, nerve granulomas or—most commonly—by granulomatous basal meningitis. Facial palsy is the most common cranial neuropathy, being unilateral in 65% and bilateral in 35%.19,20 A prominent meningitic reaction around the brainstem appears to be the underlying cause, rather than the previously assumed disease of the parotid gland—a fact supported by the lack of any temporal relation between facial nerve paralysis and parotitis, and the often abnormal brainstem auditory evoked potentials and cerebrospinal fluid (CSF) findings.21 Facial nerve palsy alone appears to be associated with a good prognosis, but other presentations of neurosarcoidosis are reported to have a poor outcome.22
The appearance of uveitis, parotid-gland enlargement, fever and cranial neuropathy (usually involving the facial nerve) is often referred to as Heerfordt’s syndrome, and is very suggestive of sarcoidosis. Interestingly, there appears to be an increased frequency of both eye and cardiac involvement in patients with neurosarcoidosis compared with other patients with sarcoidosis,23 and indeed optic nerve involvement is the second most common cranial neuropathy. Papilloedema is frequently reported and results from obstruction to CSF flow caused by chronic meningitis, or from the mass effect of large granulomata in the orbit or the cranial cavity, which may lead to optic atrophy. A subacute optic neuritis with little response to steroids and poor recovery on long-term follow-up is also seen.
In addition, syndromes deriving from the involvement of virtually every other cranial nerve have been reported.19
Epileptic seizures
These often suggest a more severe progressive or relapsing clinical course, and are most likely a reflection of underlying intracranial mass lesions, vasculopathy, or hydrocephalus.24 They are often poorly responsive to corticosteroids.22,25 In one study of 79 patients with neurosarcoidosis, 15% had seizures, and this was the first manifestation of neurosarcoidosis in 10%. All types of seizure are seen, but most are generalised tonic-clonic.25
Meningitis
Acute or chronic meningitis is seen in 3–26% of neurosarcoidosis cases. A lymphocytic CSF with raised protein levels is common, and approximately one fifth have a low CSF glucose level.26,27 Acute meningitis is usually steroid-responsive, but chronic meningitis often requires long-term steroids because of further relapses. Hydrocephalus and an “idiopathic intracranial hypertension”-like syndrome may complicate sarcoid meningitis.6,20
Mass lesions
Localised granulomatous mass lesions may affect any part of the central nervous system (CNS). These may be single or multiple and vary from small abnormalities to large tumours causing headache, lethargy and seizures.20,28,29 However, as neurosarcoidosis often has a predilection for the base of the brain, hypothalamic and pituitary gland involvement is common and may cause neuroendocrine effects with hyperprolactinaemia, the galactorrhoea-amenorrhea syndrome, hypothalamic hypothyroidism or diabetes insipidus. These features commonly present difficult diagnostic problems, especially when they are the only manifestation. Normal pituitary function tests (in particular serum prolactin levels) do not exclude hypothalamic or pituitary sarcoidosis.
Brainstem and cerebellar lesions are recognised, but are less common. Sarcoid mass lesions may also affect the optic nerve in the orbit, mimicking a meningioma, with consequent visual impairment, papilloedema and optic atrophy. Periventricular white matter lesions resembling multiple sclerosis are frequently observed on brain MRI.30
Spinal cord disease
This includes extradural, intradural or intramedullary lesions, which may be radiographically indistinguishable from a neoplastic process.31 Any segment of the cord may be involved producing various manifestations such as paraparesis, tetraparesis, radicular pain, autonomic failure, cauda equina syndrome or sphincteric disturbances. Clinically such presentations are rare and are usually preceded by a history of systemic sarcoidosis, but autopsy studies have revealed asymptomatic involvement of the spinal cord in a third of patients with sarcoidosis. There may be a CSF pleocytosis, with increased protein levels, and oligoclonal bands. Most patients presenting with spinal cord disease deteriorate significantly over 18 months or more.26,32–34
Psychiatric manifestations
These occur in about 20% of those with neurosarcoidosis, reflecting the potential for granulomatous infiltration of any part of the CNS. It is not unheard of for patients to have been committed to psychiatric care for a wide range of mental symptoms, including apathy, lack of judgement, agitation, delirium, hallucinations, irritability, lethargy and depression.35–37 Cognitive or psychiatric symptoms in a patient with multisystem sarcoidosis should therefore prompt detailed assessment because these may improve with corticosteroids.
Movement disorders
Some, such as cerebellar ataxia are commonly reported, but chorea, hemiballismus and parkinsonism are rare.26,38
Peripheral nerve and muscle disease
Peripheral neuropathies occur in approximately 15% of neurosarcoidosis patients and usually have a better prognosis than CNS involvement.26 There are various forms including axonal or demyelinating sensory and/or motor neuropathies, which may affect single or multiple nerves,26 and rarely Guillain-Barré syndrome.22,39 Asymptomatic skeletal muscle involvement is more common than symptomatic muscle disease, with the latter having nodular, acute, or more commonly chronic myopathic forms that tend to affect females in their sixth decade.26,40 Nodular myopathy is important because it may be confused with a soft tissue tumour. The response to corticosteroids is often unpredictable.41
HOW SHOULD WE MAKE THE DIAGNOSIS OF NEUROSARCOIDOSIS?
The clinical diagnosis of neurosarcoidosis is relatively straightforward in patients with multisystem sarcoidosis who later develop the typical neurological features described above. However, as early involvement of any part of the nervous system is not infrequent, this may pose a serious diagnostic challenge to the neurologist. How then should one make a diagnosis in such circumstances? (table 1). Clearly, the first step is to confirm the clinical impression of neurological involvement with targeted investigations. If these do suggest the possibility of neurosarcoidosis, the next step is a thorough search for any supportive systemic features of the disease to help secure the diagnosis.
Confirming neurosarcoidosis
Many investigations can point towards or support a diagnosis of possible neurosarcoidosis, unfortunately none short of biopsy proves it.
Brain imaging
Magnetic resonance imaging has displaced contrast enhanced CT as the most useful test for detecting basal sarcoid meningeal disease. MRI is also far superior in defining parenchymal involvement, with a reasonable level of sensitivity for intracranial abnormalities (up to 82%) but poor specificity, with a wide spectrum of imaging findings.42,43 The unenhanced MRI is unsatisfactory because of the artefacts near bone, CSF and meninges on both T1 and T2-weighted images. About 40% of patients with neurosarcoidosis have either leptomeningeal enhancement with gadolinium (fig 2), or multiple white matter lesions in a periventricular distribution that may be difficult to distinguish from those seen in multiple sclerosis.30 Infections with tuberculosis, other bacteria and fungi, and neoplastic processes such as meningeal carcinoma, lymphoma and leukaemia may also enhance with gadolinium and mimic sarcoid radiographically. The low diagnostic yield for MRI in some series may reflect the effect of therapy because the MRI changes can disappear with successful treatment of the disease. In acute myositis, MRI can show increased signal intensity in the muscles.40

Meningeal enhancement on T1-weighted MRI.
Fluorodeoxyglucose positron emission tomography (FDG-PET) may reveal areas of brain hyper- or hypometabolism, and areas of hypermetabolism outside the brain, suggesting neuro- or systemic sarcoidosis respectively.44 Although we would not routinely recommend PET scanning in suspected neurosarcoidosis, it can be helpful in supporting the diagnosis or identifying a site for potential biopsy in a puzzling case, particularly when MRI and other investigations do not provide sufficient information.45
Cerebrospinal fluid
Cerebrospinal fluid examination is helpful in the sense that it often demonstrates both a pleocytosis—predominantly a lymphocytosis ranging from 10–200 lymphocytes per cubic mm20 (but mean 24 lymphocytes from Zajicek biopsy positives cases)19—and raised protein (mean 2.7 g/l (unpublished data) and median 2.6 g/l from Zajicek biopsy positive cases). In our own study the figures were a mean of 30 lymphocytes and protein of 2 g/l (unpublished data). There may also be a high opening pressure, and in some cases a low glucose level consistent with a leptomeningitic process. Clearly such changes should also prompt CSF stains and cultures for fungi and mycobacteria.20 Patients with localised space-occupying lesions may have unremarkable CSF findings.
Some regard CSF angiotensin converting enzyme (ACE) levels as being useful in both the diagnosis and follow-up of neurosarcoidosis.46,47 However, many authorities now regard this test as non-specific and insensitive. CSF ACE may be raised in infections and malignancy, and appears unhelpful in any therapeutic decision making.48 This is illustrated by one study of 32 patients with sarcoidosis, including 20 with neurosarcoidosis: CSF ACE levels were raised in only 55% of patients with neurosarcoidosis, in 5% of patients with sarcoidosis not apparently involving the nervous system, and in 13% of patients with other neurological diseases.49
Most studies report evidence of intrathecal synthesis of immunoglobulins with oligoclonal bands in about 30%–40% of patients with neurosarcoidosis,19 while others suggest that this is uncommon.27 In one of the largest retrospective neurosarcoidosis series, all patients positive for CSF oligoclonal bands also had a raised CSF protein.19 This finding has been reproduced in our subsequent (as yet unpublished) study of 30 new cases of neurosarcoidosis. Therefore, in those band-positive patients with additional clinical or radiographic similarities to multiple sclerosis sufficient to cause diagnostic uncertainty, a raised CSF protein may contribute as a discriminant, pointing to sarcoid not multiple sclerosis (table 2).
Distinguishing between multiple sclerosis and neurosarcoidosis: in favour of neurosarcoidosis
Additional CSF markers have shown some promise but are not widely employed:
-
CSF lysosome and beta 2-microglobulin are raised in some cases and are due to local CNS secretion rather than leakage through the blood-brain barrier.50,51
-
An increased helper-suppressor T-lymphocyte ratio may help differentiate neurosarcoidosis from multiple sclerosis52,53 and in combination with raised CSF ACE, provides support for the diagnosis.
-
A CD4:CD8 ratio >5 in the CSF has been proposed as another useful diagnostic criterion for neurosarcoidosis.54
Neurophysiology
Visual, auditory or somatosensory evoked potentials can detect cranial nerve or spinal cord lesions, but their value is principally in disclosing subclinical abnormalities, rather than directly aiding differential diagnosis, for example versus multiple sclerosis.55,56 The EEG adds little diagnostic information and is often non-specifically abnormal, but it may detect the early stage of acute encephalomeningitis and epileptic discharges caused by the disease.24,57 Peripherally, electromyography and nerve conduction studies can demonstrate myopathy and large-fibre neuropathy associated with the disease. Temperature threshold testing or skin biopsy are needed to assess small-fibre neuropathy.58
Direct neural tissue biopsy
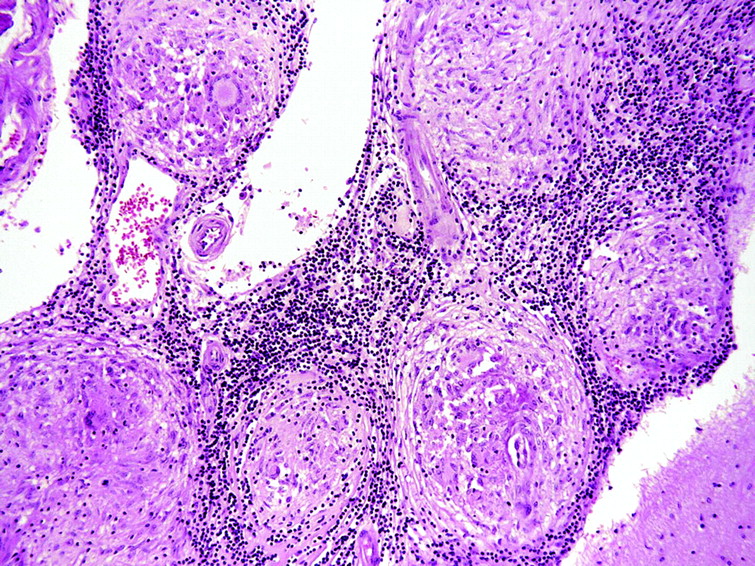
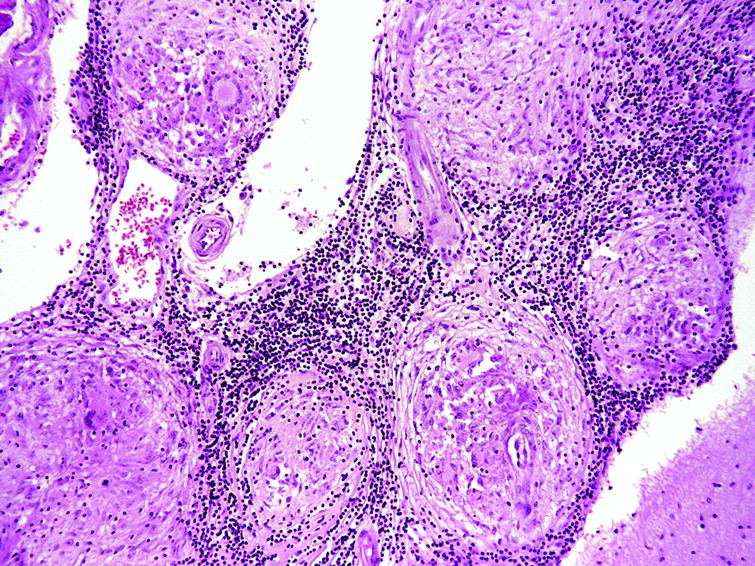
Ideally, a secure diagnosis of neurosarcoidosis requires histological evidence, with direct biopsy showing typical granulomas consisting of epithelioid cells and macrophages in the centre of non-caseating lesions surrounded by lymphocytes, plasma cells and mast cells (fig 3). In favour of doing a brain biopsy is an accessible brain lesion, little or no supportive evidence of systemic disease, and a likelihood of using immunosuppressants. Accessing cerebral tissue of course carries substantially greater risks than biopsy of other tissues, and it is not surprising that case series of neurosarcoidosis have limited numbers of patients with histological proof of the diagnosis.
{kind=link}
{kind=link}
{kind=link}
Histopathology of neurosarcoid (parietal lobe; H&E, x10).
Of course, a history of systemic sarcoidosis does not necessarily prove any new neurological problem in the patient is attributable to the disease. And even when a “positive” biopsy is obtained, further careful evaluation is needed to rule out other causes of granulomatous diseases or reactions such as infections (cryptococcus, histoplasma, toxoplasma, treponema, tuberculosis and Whipple’s disease), chemicals (silica, beryllium and contrast agents), tumours (carcinoma, lymphoma, pinealoma), inflammatory disorders (Wegener’s granulomatosis, giant cell arteritis, systemic lupus erythematosus, Churg–Strauss syndrome), and radiotherapy and chemotherapy induced changes.
DEMONSTRATING SARCOIDOSIS
Blood tests
Serum ACE levels are raised in 50% of patients with neurosarcoidosis (range 35–75%). Soluble interleukin-2R level probably has the same specificity but higher sensitivity (83%) than the serum ACE in pulmonary sarcoidosis and may be an indicator of disease progression,59 but its use in helping to diagnose neurosarcoidosis requires further evaluation. Other biochemical indices are unhelpful in establishing neurological involvement; hypercalcaemia is often seen in established pulmonary disease (a result of additional α-hydroxylation occurring in the sarcoid lesions in the lung), but many cases of neurosarcoidosis are reported with completely normal calcium levels, and similarly the erythrocyte sedimentation rate (ESR) is only raised in a minority.
Chest radiography
The diagnostic value of chest radiography in suspected systemic sarcoidosis is undisputed: abnormalities are seen in 90%, but of course, a normal chest x ray at presentation does not exclude generalised sarcoidosis.14 Most commonly observed is bilateral hilar lymphadenopathy, which is usually asymptomatic and subsides without treatment in 80–90% of patients. Less common are pulmonary infiltrations with symptoms of dyspnoea, cough and fever that may also resolve, or pulmonary fibrosis leading to permanent bullae formation, associated with restrictive lung defects. Chest x ray abnormalities in neurosarcoidosis are variable, with a reported frequency ranging from 31%–80%.19,22 This clearly means that one cannot be reassured by a normal chest x ray when considering a diagnosis of neurosarcoidosis.
High definition CT thorax
This is useful in diagnosing pulmonary disease and also assists in targeting transbronchial biopsy. It is approximately 89% and 98% sensitive for hilar and mediastinal adenopathy respectively, and 23% sensitive for pulmonary infiltrates.60 High definition CT and bronchoalveolar lavage with immunocytochemistry showing a CD4:CD8 ratio >3.5 has been proposed as an additional diagnostic criterion of neurosarcoidosis,54 though there have been no exhaustive studies of sensitivity and specificity.
Gallium scanning
Whole body gallium scanning, although of relatively low specificity, has a sensitivity of 60–90%43 and is a useful indicator of systemic disease. Ga-67 citrate is taken up at sites of active sarcoidosis, but also by other inflammatory and malignant diseases, including tuberculosis and lymphomas. Although a typical pattern of uptake in the salivary glands, chest and muscle is well recognised among patients with active sarcoidosis, increased uptake in the cranium is reported relatively infrequently.40,61,62
Kveim–Siltzbach test
This test is now largely only of historical value because it is rarely available, but its notable success in contributing to the diagnosis of neurosarcoidosis deserves special mention. Material for the test is obtained from human sarcoid spleen and is injected intradermally. In patients with active sarcoidosis, epithelioid cell granulomata gradually develop and can be demonstrated following biopsy of the purplish-red nodule that is formed. The test is highly sensitive for neurosarcoidosis, positive in up to 85% of cases—only slightly less sensitive than direct positive histology from tissue outside the affected nervous system.19 However, other than the potential for the transfer of infection there are two distinct disadvantages of this test: 4–6 weeks must elapse before a biopsy can be performed, which may be a crucial delay, and steroids may make the test negative.
Systemic (“blind”) organ tissue biopsy
Biopsy of a lymph node, lung, liver, skin or conjunctiva may be considered if involvement is suspected following clinical and/or radiological assessment.20,63–65 And even without apparent involvement, such biopsies may still be performed “blind” when there is suspicion of neurosarcoidosis but no systemic clinical abnormality. An example is asymptomatic muscle involvement, which occurs in 50–80% of patients or more.66 A generous muscle biopsy with multiple sectioning could therefore help to confirm the presence of typical granulomas surrounded by normal muscle: gastrocnemius, vastus lateralis or brachial biceps are possible target muscles.67
How certain can we be of the diagnosis of neurosarcoidosis?
Although the criteria on which a clinical diagnosis of neurosarcoidosis is made in the absence of positive nervous tissue histology have not been firmly established, one generally requires a clinically compatible picture, exclusion of other neurological disease, and histological confirmation of sarcoid elsewhere. Zajicek and colleagues19 formulated a series of diagnostic criteria in 1999 to include biochemical abnormalities and important diagnostic techniques such as MRI, gallium scanning, chest x ray, CSF and the Kveim–Siltzbach test, which help to define probable and possible disease. Others have since refined these criteria to exclude the Kveim–Stilbach test (which as already mentioned is no longer used), chest x ray and serum ACE (which as described above are relatively poor markers of CNS disease). Instead they have included high definition CT of the chest and bronchoalveolar lavage with a CD4:CD8 ratio >3.5, and a CD4:CD8 ratio >5 in the CSF.54 Table 1 incorporates these modifications to provide reasonable diagnostic criteria.
TREATMENT
Considering the rarity of neurosarcoidosis and the difficulties in diagnosis, it is unsurprising to find there are no randomised controlled trials of treatment. Recommendations are therefore based on retrospective case series, anecdotal experience, and randomised controlled trials in pulmonary sarcoidosis. It is generally accepted that treatment is difficult, and this is reflected by the significant associated morbidity and mortality from the disease.
Corticosteroids remain the cornerstone of therapy, but adverse effects are prominent, because the required dosage can be high, with the frequent need for prolonged therapy.19,22,68–70 The daily dose of prednisolone varies from 40–80 mg, but the symptoms tend to recur at less than 20 mg/day, making withdrawal difficult.6 Any concomitant antiepileptic drug that induces hepatic microsomal enzymes may reduce prednisolone concentration and efficacy, necessitating even higher oral doses. Bolus-pulsed intravenous methylprednisolone gives a high initial loading dose of corticosteroid and may help to avoid the adverse effects associated with long-term oral treatment. Treatment is therefore often initiated with 1 g of intravenous methyl prednisolone daily for three days followed by 0.5–1 mg/kg oral prednisolone per day. An enhanced MRI brain scan at this stage may be useful—persistent or new enhancement suggests that further reduction in corticosteroid therapy may lead to an exacerbation of the disease.71 Conversely, persistent CSF abnormalities do not necessarily correlate with disability and are therefore not an indication for continuing therapy.6 If the disease does become quiescent on a low dose of prednisolone, the daily dose can be further tapered by 1 mg every 2–4 weeks.6 Methotrexate or hydroxychloroquine may then be added, especially if the response to steroids is inadequate.23,69
Methotrexate used weekly at an oral dose of 10 mg, is well tolerated with minimal adverse effects and may be of value in maintaining optimal disease suppression together with intravenous or oral steroids.19 It is recommended that patients undergo a liver biopsy after every gram of methotrexate administered, but irreversible liver damage is fortunately rare.
Chloroquine has now largely been replaced by the less toxic hydroxychloroquine at a dose of 200 mg/day, and may be used for up to one year as a first line agent with methotrexate. Hydroxychloroquine is also particularly effective in patients with skin lesions, and its hypoglycaemic effect may be useful in those with steroid-induced hyperglycaemia.
Few data are available to support the use of other forms of immunotherapy in neurosarcoidosis. Chlorambucil, cyclophosphamide, cyclosporin, azathioprine and mycophenolate mofetil have all been used, with variable results,6,19,68,72,73 usually in combination with corticosteroids which can rarely be withdrawn altogether—the best combination being a moderate dose of prednisolone with an adjunct therapy.6 One study of cyclosporin treatment in neurosarcoidosis showed only a modest response and allowed a mere 30–55% reduction in the steroid dosage.72 Cyclophosphamide is often considered, several studies reporting significant neurological improvement.23,68,74,75 Compared with oral treatment, pulsed intravenous cyclophosphamide is associated with better patient compliance and lower risk of malignancy, especially bladder cancer.
Although the chemokine and cytokine pathways that regulate granuloma formation are not well understood, tumour necrosis factor-alpha (TNF-α) is implicated.76 The TNF-α antagonists pentoxifylline and thalidomide are reported to be useful in refractory systemic and neurosarcoidosis.77,78Infliximab (a monoclonal antibody against TNF-α) in particular has a growing body of literature supporting its effectiveness, and appears to be a safe treatment with good steroid sparing effects.79–83
Practice points
-
Although sarcoidosis is rarely confined to the nervous system, its neurological features frequently occur early in the course of the disease leading to diagnostic confusion.
-
Presentation with cranial neuropathies (particularly nerves VII, II) is the most common, but seizures, chronic meningitis and the effects of mass lesions are also frequent manifestations of neurosarcoidosis.
-
The diagnostic process should first confirm nervous system involvement and then provide supportive evidence for systemic disease.
-
In the absence of direct tissue biopsy, the most useful diagnostic tests for neurosarcoidosis are gadolinium enhanced MRI of the brain and CSF analysis.
-
The diagnostic criteria for neurosarcoidosis have been modified to include high definition CT of the chest and bronchoalveolar lavage with a CD4:CD8 ratio >3.5, and a CD4:CD8 ratio >5 in the CSF.
-
The mainstay of treatment is corticosteroids, which often need to be combined with other immunosuppressants.
The long-term complications of these cytotoxic agents should not be underestimated, with awareness for the effects of immunocompromise. Infections such as toxoplasmosis, tuberculous and cryptococcal meningitis may occur, and also lymphoproliferative disorders, which may be overlooked as some features of these diseases may themselves mimic sarcoidosis.
Antiepileptic drugs are indicated for seizures associated with the disease. Depression is common and appropriate antidepressant therapy should not be withheld. Phenytoin (and also methotrexate) may cause hilar and mediastinal lymphadenopathy, which may be indistinguishable from sarcoidosis.
Radiotherapy is a treatment option usually limited to those patients whose symptoms cannot be controlled on steroids and/or cytotoxic drugs, or for those who cannot tolerate their adverse effects.6 The mechanism can only be extrapolated from our knowledge of the radiobiological effects on normal and neoplastic tissue: metabolically active cells of granulomata are probably more susceptible to radiation damage than normal CNS tissue. Case reports have shown variable clinical and radiological outcomes.84–86
Neurosurgical intervention is indicated in life-threatening situations or when medical treatment fails. Asymptomatic ventricular enlargement does not usually require surgery, and mild symptomatic hydrocephalus may simply respond to corticosteroids.15 More significant hydrocephalus will require a CSF diversion procedure, usually ventriculoperitoneal shunting,87 although endoscopic treatment may be successful.88 Shunt obstruction may occur in these patients by inflamed CSF, ependyma or rarely non-caseating granulomas.89 Resection of intracranial or spinal granulomata may be required, especially when they result in raised intracranial pressure.
Acknowledgments
The authors are grateful to Professor Seth Love and Dr Marcus Likeman for the kind provision of illustrative material. This article was reviewed by Myles Connor, Edinburgh, UK.
REFERENCES
Linked Articles
- From the editor's desk
Other content recommended for you
- Investigating chronic meningitis
- Neurosarcoidosis: a study of 30 new cases
- Measurement of cerebrospinal fluid ACE level in aseptic meningitis: diagnostic?
- Orbitopalpebral and ocular sarcoidosis: what does the ophthalmologist need to know
- Neurosarcoidosis: clinical manifestations, investigation and treatment
- Bell’s that did not ring true
- Aseptic meningitis and hydrocephalus secondary to neurosarcoidosis
- Hydrocephalus as the first presenting symptom of neurosarcoidosis in two patients: a diagnosis more forthcoming in the context of systemic disease
- A case of neurosarcoidosis that presented with symptoms of Guillain–Barré syndrome
- Neurosarcoidosis presenting with a partial Claude syndrome