Article Text

Abstract
Neurologists should be able to anticipate and recognise the onset of respiratory failure in patients with neuromuscular disorders. Symptoms will differ depending on the speed of onset of the respiratory muscle weakness. Careful monitoring of respiratory function is particularly important in acute disorders such as Guillain-Barré syndrome. Patients with an unrecognised neuromuscular disorder may occasionally present with respiratory failure. Important investigations include vital capacity, mouth pressures, arterial blood gases, chest x ray and sometimes overnight respiratory monitoring. Patients with Guillain-Barré and other acute conditions may require short-term ventilatory support in the intensive care unit. Patients with some chronic disorders, such as motor neuron disease and Duchenne dystrophy, can be successfully treated with non-invasive ventilation, usually in collaboration with a respiratory physician. New-onset weakness of limb and respiratory muscles in the intensive care unit is usually due to critical illness myopathy or critical illness polyneuropathy, and treatment is supportive.
Statistics from Altmetric.com

Respiratory failure occurs when the movement of air in and out of the lungs is compromised by weakness of the respiratory muscles. This may arise acutely in conditions such as Guillain-Barré syndrome (GBS). More gradual-onset respiratory failure occurs in patients with chronic neuromuscular disorders. Neurologists should be able to anticipate and recognise the onset of respiratory failure in both groups of patients. Rarely, the neurologist may encounter a patient with an unrecognised neuromuscular disorder who has presented de novo with acute-on-chronic respiratory failure. The aim of this article is to review the presenting symptoms and signs of respiratory failure, which neuromuscular disorders are particularly associated with respiratory muscle weakness, and the respiratory investigations which the neurologist should know about.
NORMAL PHYSIOLOGY
The diaphragm is the most important inspiratory muscle, responsible for approximately 70% of ventilation at rest. Accessory muscles of inspiration include the external intercostal, scalene and sternocleidomastoid muscles. Muscles of expiration include the internal intercostal and abdominal wall muscles; these are not required during breathing at rest, due to the passive recoil properties of the thoracic cage, but are important for the generation of an adequate cough.
As with any organ, there is substantial reserve in the respiratory system. Assuming normal function of the brain and lungs, respiratory failure will not usually occur until respiratory muscle strength has fallen to around 25–30% of normal. The accessory muscles can provide adequate ventilation in patients even with diaphragmatic paralysis from bilateral phrenic nerve palsy, and around half the patients with unilateral phrenic nerve palsy are entirely asymptomatic.1
PATHOPHYSIOLOGY
In addition to reduction of airflow, respiratory muscle weakness results in a decline of the functional residual capacity of the lungs. This increases the work of breathing, because at lower volumes the lungs are less distensible. Further, the reduction of lung volume alters ventilation/perfusion relationships, resulting in less efficient gas exchange.
The adequacy of central respiratory drive is particularly important in patients with respiratory muscle weakness. In normal rapid eye movement (REM) sleep, there is transient reduction of the respiratory drive, together with hypotonia of the accessory respiratory muscles. Therefore, patients with subacute or chronic respiratory muscle weakness will usually develop hypopnoea, oxygen desaturation and hypercarbia for the first time during REM sleep.
Once respiratory reserve is compromised, any increase in the respiratory load can lead to diaphragmatic fatigue and respiratory failure. Factors which increase the load include an increase in the respiratory rate (for example, with fever), an increase in stiffness of the lungs (for example, lung consolidation or atelectasis), and abdominal distension (for example, constipation).
SYMPTOMS
The symptoms of respiratory muscle weakness depend on the speed of its development:
When the onset is subacute (for example, GBS) the predominant symptoms are dyspnoea and orthopnoea, or sometimes respiratory arrest. These symptoms are often accompanied by those of bulbar weakness and inability to clear respiratory secretions. The symptoms of respiratory failure may easily be overlooked and should be specifically sought in any patient with rapidly progressive weakness, especially when the bulbar muscles and shoulder girdle are affected.
When respiratory muscle weakness develops gradually, inadequate respiration usually occurs first during sleep. Symptoms of nocturnal hypoventilation include a broken sleep pattern, nightmares, nocturnal confusion, morning headache, daytime fatigue, mental clouding and somnolence.
Exertional dyspnoea is encountered less frequently in neuromuscular patients than in those with other cardiorespiratory disorders, particularly when the patient has reduced mobility. Dyspnoea when lying flat or immersed in water specifically suggests weakness of the diaphragm.
SIGNS
A patient with severe respiratory muscle weakness or respiratory failure may appear overtly breathless and be using accessory muscles of respiration. He or she may be unable to speak in complete sentences or take deep breaths to command. Inability to count from 1 to 20 in a single breath indicates significant reduction of vital capacity. Paradoxical abdominal motion (inwards movement of the abdominal wall with inspiration) suggests significant weakness of the diaphragm. The combination of hypoxaemia and hypercarbia may produce mental clouding or somnolence.
It is also important to assess the bulbar musculature, weakness of which can hinder clearing of respiratory secretions and so allow aspiration. Acute respiratory failure in patients with neuromuscular disorders is often precipitated by respiratory infection.
Most patients with respiratory muscle weakness resulting from a neuromuscular condition (table) have limb weakness and other signs of that disorder, familiar to neurologists. Particular attention should be paid to:
presence or absence of bulbar weakness
a tall, thin face (congenital myopathy, myotonic dystrophy)
ptosis or ophthalmoparesis (mitochondrial disorder, myasthenia)
fasciculation (motor neuron disease)
paraspinal muscle wasting (acid maltase deficiency)
skin rash (dermatomyositis).
RESPIRATORY INVESTIGATIONS
Neurologists should have some familiarity with the following respiratory investigations:
Vital capacity (VC) is a simple, widely available respiratory function test. Normal values are calculated from the patient’s age, height and sex. In patients with weakness of facial muscles it is important to ensure a good seal between the mouthpiece and the lips. A fall in the VC by more than 15–20% when the patient lies down specifically suggests weakness of the diaphragm. A normal VC, with no significant fall when supine, means that respiratory muscle weakness is unlikely to be responsible for a patient’s respiratory symptoms. However, bear in mind that the muscle weakness in myasthenia gravis can fluctuate significantly.
Mouth pressures. The maximal inspiratory pressure (MIP) can further assess the strength of respiratory muscles. It is usually measured in the respiratory laboratory using the same equipment which records the VC. It critically depends on maximal effort from the patient. If the MIP is low, the sniff nasal inspiratory pressure (SNIP) can be checked and the higher of the two values used. In an adult male, an MIP of more than 80 cm H2O (female: 70 cm H2O) or a SNIP of more than 70 cm H2O (female: 60 cm H2O) excludes clinically significant respiratory muscle weakness.
Peak cough flow can be measured at the bedside with a standard peak flow meter. It should be viewed as ancillary to the VC. The normal range in adults is 360–840 l/min. A value <160–200 l/min may be associated with inability to adequately clear the respiratory tree of secretions, and suggests that assisted coughing techniques are indicated.
Arterial blood gases. Derangement of arterial blood gases is a late feature in neuromuscular disease and normal results are quite compatible with significant weakness of the respiratory muscles. Patients with established respiratory failure from neuromuscular weakness will show hypoxaemia and a compensated respiratory acidosis (raised PaCO2 and bicarbonate with a normal or mildly reduced pH). Elevations of the pH and bicarbonate with normal PaO2 and PaCO2 suggest nocturnal hypoventilation.
Chest x ray provides essential information about the presence or absence of lung infection, atelectasis, or kyphoscoliosis. A unilateral phrenic nerve palsy is suggested by a raised hemidiaphragm; this can be confirmed by fluoroscopy or ultrasound while the patient performs a sniff (a paralysed or very weak diaphragm will ascend rather than descend).
Overnight monitoring. In neuromuscular patients, alveolar hypoventilation during sleep typically occurs prior to frank respiratory failure. The most straightforward overnight investigation in a patient suspected to have alveolar hypoventilation is oximetry, with transcutaneous PaCO2 recording if available. This will usually be performed in a Respiratory Department or Sleep Laboratory. Where available, polysomnography can also be considered, to provide simultaneous monitoring of heart rate, ventilatory effort, airflow, electroencephalogram, eye movements, oxygenation and muscle tone.
MONITORING PATIENTS WITH AN ACUTE NEUROMUSCULAR DISORDER
Guillain-Barré syndrome
Approximately 25% of patients with GBS require intubation and ventilation, usually due to a combination of bulbar and respiratory muscle weakness. In different studies, the following features have been found to predict the need for ventilation:2–4
time from onset of symptoms to admission <7 days
rapid progression of disability
bulbar dysfunction
bilateral facial weakness
inability to stand
inability to lift head or elbow off bed
dysautonomia
vital capacity <20 ml/kg
maximum inspiratory pressure (MIP) <30 cm H2O
maximum expiratory pressure (MEP) <40 cm H2O
serial decline of the vital capacity, MIP or MEP by >30%
demyelination on neurophysiological testing
raised liver enzymes.
In patients in the deterioration phase of GBS, repeated assessment is important. Particular attention should be directed to the status of the bulbar and respiratory muscles. The vital capacity and oxygen saturation should be rechecked at frequent intervals. A vital capacity <1 litre in an adult (more accurately, <15 ml/kg), a fall in the vital capacity by more than 50% on serial testing, or onset of bulbar palsy are all indications to involve the intensive care unit.
Poliomyelitis
Acute poliomyelitis typically presents with with fever, headache, neck stiffness and asymmetric flaccid paralysis without sensory loss. Respiratory involvement occurs in about 15% of patients. There is a pleocytosis in the cerebrospinal fluid, and MRI may show abnormal signal involving the anterior horns of the spinal cord. As it is now rarely seen in many countries, poliomyelitis can initially be misdiagnosed as GBS or transverse myelitis. Respiratory monitoring should be the same as for GBS.
Other acute neuromuscular disorders include porphyria, organophosphate poisoning, myasthenia gravis, botulism and rhabdomyolysis.
MONITORING PATIENTS WITH A CHRONIC NEUROMUSCULAR DISORDER
Any generalised neuromuscular disorder which becomes severe can theoretically involve the respiratory muscles. In practice, only a limited set of neuromuscular conditions is accompanied by respiratory failure with appreciable frequency (table). Patients who could potentially develop respiratory muscle weakness should have their vital capacity and mouth pressures measured at least once. Whether to repeat the testing will depend on the diagnosis and the rate of progression of muscle weakness. A serial fall in the vital capacity (particularly a fall below 1.2–1.5 l or <40–50% of predicted), or a fall in MIP to <60% of predicted, are indications for further respiratory assessment. Onset of daytime hypercapnia (PaCO2 >45 mmHg or 6 kPa) indicate that the patient needs urgent assessment and may imminently require non-invasive ventilation.
Motor neuron disease
All neurologists are familiar with the clinical features of this disorder. Presentation with respiratory muscle weakness or with hypercapnic respiratory failure is rare but well-recognised. Whatever the mode of onset of their disease, most patients eventually develop respiratory muscle weakness, and this can be anticipated by serial measurement of the VC and MIP. Symptoms include exertional dyspnoea, orthopnoea, fatigue, non-refreshing sleep and morning headache. There is good evidence that non-invasive ventilation can improve these symptoms and quality of life. It also significantly improves survival (to a much greater extent than does riluzole) in those patients with only mild or moderately impaired bulbar function.5 Where resources are available, a respiratory physician with an interest in non-invasive ventilation should be part of the management team in motor neuron disease (see below).
Spinal muscular atrophy
Types 1, 2 and 3 are recognised, depending on age of onset. All forms have the same genetic basis—homozygous deletion of the telomeric survival motor neuron gene. Respiratory muscle involvment in spinal muscular atrophy type 1 (Werdnig-Hoffman disease) is universal. It occurs to a variable extent in Type 2, and is uncommon in Type 3.
Congenital myopathy
In the congenital myopathies, onset of hypotonia and weakness occur in early life. Unlike congenital muscular dystrophy, distinct structural alterations (for example, rod bodies in nemaline myopathy) are apparent within the muscle fibres at biopsy. If the child survives infancy, there is usually minimal or no progression of weakness in later life.
Of the more than 12 well-characterised congenital myopathies, respiratory muscle involvement is best recognised in nemaline myopathy (fig 1), myotubular (centronuclear) myopathy and multiminicore disease. If it occurs at all, respiratory failure is usually evident in the neonatal period. However, in a rare variant of nemaline myopathy—sporadic late-onset nemaline myopathy—the onset of limb and respiratory muscle weakness occurs in adult life.6

Congenital muscular dystrophy
In this rare set of disorders, hypotonia, weakness, and contractures occur within the first six months of life. In some forms there may be structural brain disease and impaired cognitive function. Respiratory muscle involvement and respiratory failure are common features and often prove fatal in childhood or the second decade.
Dystrophinopathy
Duchenne muscular dystrophy and its less severe allelic counterpart, Becker muscular dystrophy, arise from mutations of the gene encoding dystrophin. In Duchenne, weakness of the respiratory muscles is universal. After the normal rise in childhood, the vital capacity begins to decline after age 10. The assessment of respiratory function may be complicated by scoliosis, and by cardiac dysfunction. Non-invasive ventilation (fig 2) has been one factor which has increased the survival beyond age 24 to 53% in one large centre.7 In Becker muscular dystrophy, respiratory failure is less common but lung function should be checked at intervals.

Myotonic dystrophy
Myotonic dystrophy (DM1) results from a CTG expansion in the gene encoding DMPK. It is a multisystem disorder, but the clinical features are highly variable in different patients. Respiratory failure, pneumonia or cardiac conduction defects are the usual causes of death. As well as weakness of the respiratory muscles, patients can have a central defect of respiratory control, and be predisposed to respiratory infection due to bulbar weakness. Monitoring should include periodic checks on the vital capacity, and an annual ECG as the PR interval and QRS duration each increase by a few percent per year. The alterations of personality and cognitive function found in patients with DM1 can hinder compliance with non-invasive ventilation.
Limb girdle muscular dystrophy
Limb girdle muscular dystrophy is a steadily expanding set of autosomal dominant or recessive myopathies which have in common weakness of the shoulder and pelvic girdle muscles. Types 1A–1G (autosomal dominant) and 2A–2L (recessive) are currently recognised.
Respiratory muscle weakness has been particularly associated with types 2C, 2D, 2E and 2F which are due to mutation of the genes encoding the γ-, α-, β- and δ-sarcoglycans respectively (sarcoglycanopathies). These disorders typically present in late childhood with pelvic, then scapular, deltoid, trunk and thigh muscle weakness. Calf hypertrophy and eventual inability to walk are common.
Limb girdle muscular dystrophy 2I results from mutations of FKRP, the gene encoding fukutin-related protein. Presentation is usually in the second decade with a picture resembling dystrophinopathy: proximal weakness, calf hypertrophy and exaggerated lumbar lordosis. Respiratory muscle involvement occurred in 10 of 16 patients in one series,8 with non-invasive ventilation required in five patients.
Mitochondrial myopathy
That disease at mitochondrial level can lead to dysfunction at all levels of the nervous system will be familiar to neurologists. It is now known that mutations of both mitochondrial (mtDNA) and nuclear DNA can produce mitochondrial disorders. Respiratory involvement is well-recognised in mitochondrial myopathy, and is occasionally the presenting feature.9
Chronic progressive external ophthalmoplegia is the most common mitochondrial myopathy in our experience. It usually presents with longstanding ptosis, ophthalmoparesis and proximal limb weakness (fig 3). Laboratory features include a raised resting venous lactate, and the presence on muscle biopsy of ragged-red and cytochrome c oxidase-negative muscle fibres (fig 4).

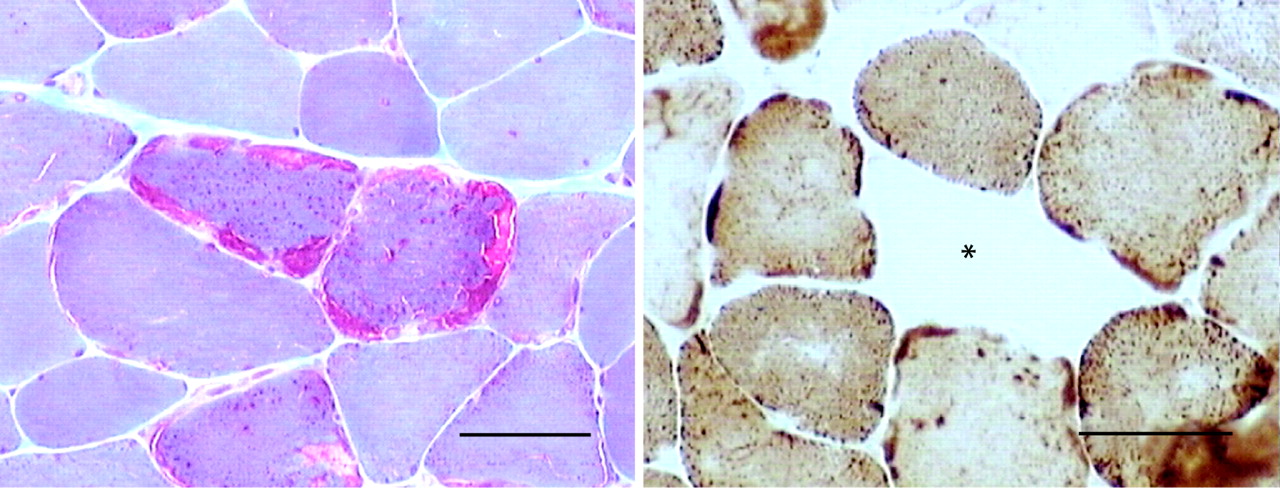
Poliomyelitis
Patients who have previously had acute polio with respiratory involvement are at risk of deterioration of respiratory function later in life (post-polio syndrome). Postulated mechanisms include loss of respiratory anterior horn cells from a small surviving pool, and failure of motor axon sprouts in massively reinnervated motor units.
Acid maltase deficiency
In acid maltase deficiency (glycogen storage disorder type II), deficiency of the lysosomal enzyme acid α-glucosidase leads to intracellular accumulation of glycogen. There are three forms:
In the infantile form, there is particular involvement of skeletal muscle, heart and liver, with death usually before age 2 from respiratory insufficiency.
The childhood form usually presents in the first years of life with proximal limb or respiratory muscle weakness.
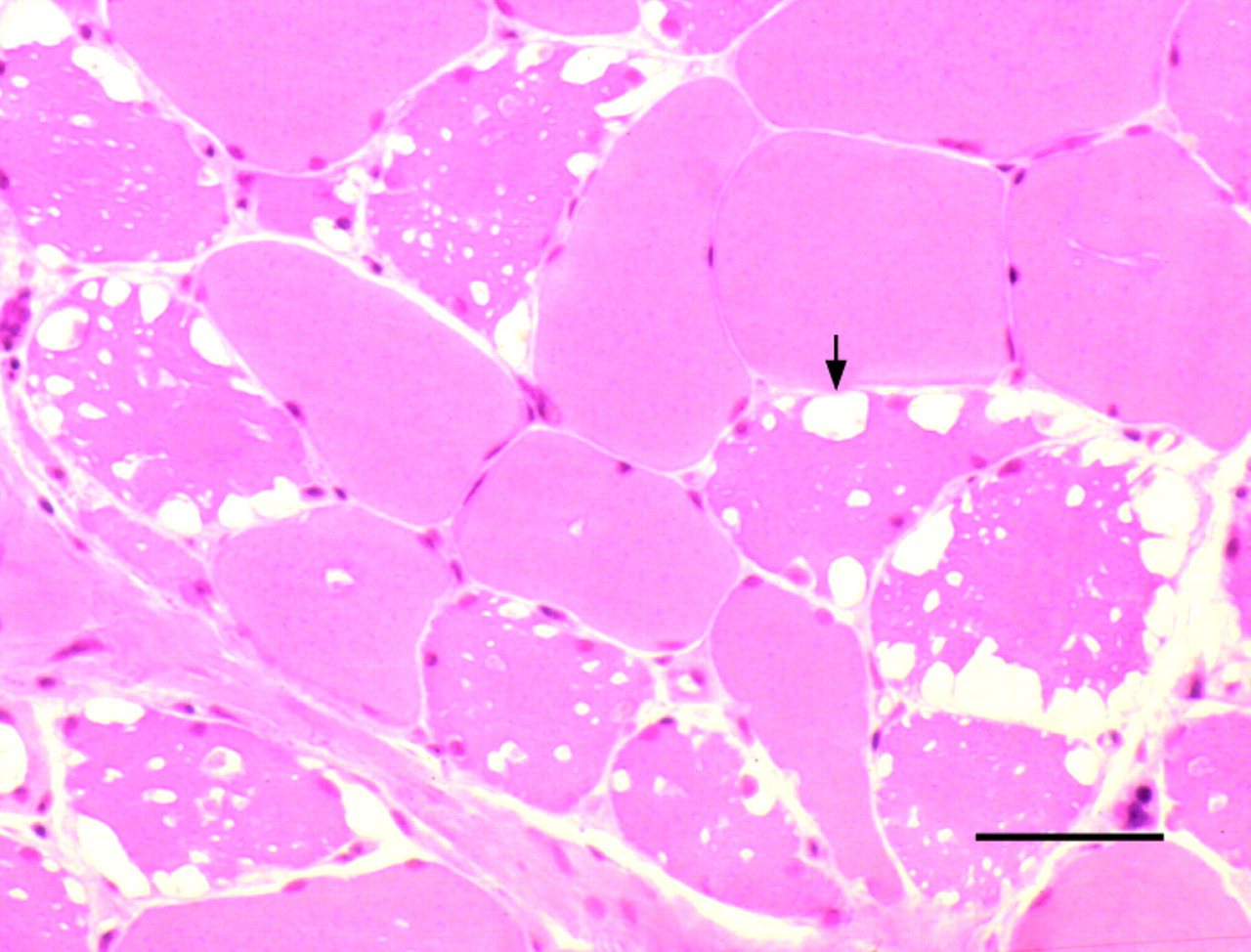
In the adult form, presentation occurs beyond childhood, usually with an indolent axial and proximal myopathy. Involvement of respiratory muscles is common, and de novo presentation with acute-on-chronic respiratory failure is well-recognised.10 Electromyography usually shows myotonic or complex repetitive discharges, although these abnormalities may be confined to axial muscles. The pathological hallmark is a vacuolar myopathy (fig 5), but there may be no characteristic histological features in clinically normal or only mildly affected muscles. The diagnosis can be confirmed by enzyme assay using a muscle homogenate, blood spot, leucocytes or cultured fibroblasts.
Myasthenia gravis
Weakness of the bulbar or respiratory muscles is the most serious complication of myasthenia gravis. Respiratory failure with hypoventilation and fatal hypercarbia can sometimes develop over hours, often in the setting of a respiratory infection. Patients who complain of dyspnoea, particularly at rest, or those experiencing some other exacerbation of their myasthenia require careful assessment of their respiratory function.
Significant respiratory muscle weakness may also occur in other disorders of neuromuscular transmission: Lambert-Eaton myasthenic syndrome, botulism and organophosphate poisoning.
PATIENTS WITH A NEUROMUSCULAR DISORDER WHO PRESENT WITH RESPIRATORY MUSCLE WEAKNESS
Presentation with respiratory muscle weakness is well-recognised in motor neuron disease, myasthenia gravis, and adult-onset acid maltase deficiency, and has rarely been reported in mitochondrial myopathy,9 sporadic late-onset nemaline myopathy6 and colchicine myopathy.11 Some of these patients will have required emergency intubation and ventilation before the involvement of a neurologist. They will almost always have peripheral signs of a neuromuscular disorder and their work-up should proceed along conventional lines.
NEW ONSET RESPIRATORY MUSCLE WEAKNESS IN THE INTENSIVE CARE UNIT
Critical illness myopathy
This condition, also called acute quadriplegic myopathy, is an acute, diffuse myopathy usually encountered in the intensive care unit (ICU). Risk factors include a critical illness (often with sepsis and multi-organ dysfunction) and administration of corticosteroids or non-depolarising neuromuscular blocking agents. The presenting symptom is often failure to wean from the ventilator. Examination shows diffuse weakness of proximal and distal muscles. Frequently the tendon reflexes are reduced. Unlike rhabdomyolysis, which can also occur in critically ill patients, the creatine kinase is normal or only modestly elevated. In part, the weakness can be attributed to muscle fibre atrophy resulting from selective loss of myosin filaments from the sarcomere, and other pathological alterations (fig 6). There is also reduced electrical excitability of muscle fibres, the result of depolarisation of the resting membrane potential and a hyperpolarising shift in the voltage dependence of inactivation of sodium channels.12

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Critical illness polyneuropathy
This is the chief differential of critical illness myopathy and the two conditions may coexist. It is particularly frequent in ICU patients who develop sepsis and multi-organ failure. If sufficiently severe, there may be failure to wean from the ventilator. The clinical features otherwise include distal-predominant weakness, reduced or absent reflexes, and sensory loss. Neurophysiological tests may or may not distinguish this from critical illness myopathy with confidence. Among other factors, tissue oedema may complicate the recording of sensory action potentials, cooperation for electromyography in an ICU patient may be limited, and the two conditions may coexist. Muscle biopsy to distinguish one from the other is seldom indicated, because the treatment of both conditions is supportive. There is usually slow recovery over months in those patients who survive their stay in the ICU.
LONG-TERM MANAGEMENT OF RESPIRATORY FAILURE IN NEUROMUSCULAR DISORDERS
The management of respiratory failure is beyond the scope of this article. Neuromuscular patients with symptomatic respiratory muscle weakness should be referred to a respiratory physician with a special interest in the management of such patients. Treatment can include respiratory muscle training, assisted cough techniques, and non-invasive ventilation. Among other factors, the decision to begin non-invasive ventilation is based on the underlying disease, its rate of progression, and the impact of the patient’s symptoms on quality of life. It places significant demands on the patient and carers, and requires motivation.
Practice points
Respiratory failure may result when the movement of air in and out of the lungs is compromised by weakness of the respiratory muscles.
Neurologists should be able to anticipate the onset of respiratory failure in patients with acute and chronic neuromuscular disorders.
Assuming normal function of the brain and lungs, respiratory failure does not usually occur until respiratory muscle strength has fallen to around 25–30% of normal.
Patients with subacute or chronic respiratory muscle weakness will usually develop hypopnoea, oxygen desaturation and hypercarbia for the first time during REM sleep.
In patients with respiratory muscle weakness, it is also important to assess the bulbar musculature.
A normal vital capacity, with no significant fall when supine, means that respiratory muscle weakness is unlikely to be responsible for a patient’s respiratory symptoms.
On arterial blood gas analysis, elevations of the pH and bicarbonate with normal PaO2 and PaCO2 suggest nocturnal hypoventilation.
In Guillain-Barré syndrome, a vital capacity <1 litre, a fall in the vital capacity by more than 50% on serial testing, or onset of bulbar palsy are all indications to involve the intensive care unit.
Patients with a chronic neuromuscular disorder who could potentially develop respiratory muscle weakness should have their vital capacity and mouth pressures measured at least once.
In a patient with a chronic neuromuscular disorder, onset of daytime hypercapnia (PaCO2 >45 mmHg or 6 kPa) indicates that the patient needs urgent assessment and may imminently require non-invasive ventilation.
In patients with motor neuron disease and respiratory failure, there is good evidence that non-invasive ventilation can improve symptoms, quality of life and survival.
De novo presentation with respiratory muscle weakness is well-recognised in motor neuron disease, myasthenia gravis, and adult-onset acid maltase deficiency.
ACKNOWLEDGEMENTS
This article was reviewed by Richard Hughes, London, UK.
REFERENCES
Footnotes
Patient consent: Obtained.
Other content recommended for you
- Weakness in the intensive care unit
- Respiratory involvement in inherited primary muscle conditions
- Muscle diseases: mimics and chameleons
- Respiratory aspects of neurological disease
- Long term ventilation in neurogenic respiratory failure
- Weakness on the intensive care unit
- Respiratory management of motor neurone disease: a review of current practice and new developments
- Orthopnoea in a young woman
- Muscle disease
- British Thoracic Society guideline for respiratory management of children with neuromuscular weakness