Article Text

Abstract
Patients who are recovering from critical illness may be weak and difficult to wean from ventilatory support as a complication of their underlying disorder, intercurrent events or treatment given during prolonged intensive care. These patients are difficult to assess because of the severity of their weakness and any accompanying encephalopathy. It is essential to undertake a meticulous review, including assessment of any septic, hypoxic or metabolic derangements and a detailed look at the dosage and duration of medication including antibiotics, neuromuscular junction blocking agents and sedation. If a primary underlying neurological cause or an intercurrent event have been excluded, the likeliest cause of weakness is one of the neuromuscular complications of critical care such as: critical care polyneuropathy, an acute axonal neuropathy which develops in patients with preceding sepsis or multi-organ failure; the use of neuromuscular junction blocking agents or steroids; and critical illness myopathy, which is the most common cause of critical care related weakness.
Statistics from Altmetric.com

Neurologists may be called to see patients on the intensive care unit (ICU) because of “weakness” which becomes apparent during or immediately following resuscitation and stabilisation. More commonly, patients are referred because they have failed to wean from ventilatory support after a severe illness or routine anaesthetic; because they have remained weak despite having been weaned from ventilation; or because they have a known primary neurological disorder and require further assessment.
ASSESSMENT
The assessment of the “weak” patient on the ICU may be difficult because:
The patient is often highly anxious or frightened by a sudden deterioration in their wellbeing and the urgent resuscitation measures undertaken by paramedics and A&E staff.
Those who have been on the ICU for a long time frequently become withdrawn because of anxiety or depression.
Obtundation or encephalopathy may occur as a consequence of the underlying disease process, its systemic complications or treatment.
As a consequence of prolonged ICU stay, the patient may have become disorientated and confused by pain, medication, intercurrent events, day/night reversal, nocturnal hallucinations and lack of sleep.
Communication may be impaired by ocular, facial, bulbar, limb or respiratory muscle weakness.
Patients who have been intubated but are not sedated also find communication stressful and exhausting.
Regardless of whether the presentation is acute, or weakness has developed after a prolonged ICU admission, there are a number of fundamental management issues.
Communication
The first priority is to establish a means of communication with the patient, a painstaking process requiring the support of intensivists, nursing staff and, most importantly, relatives and friends. Many weak patients are effectively “locked in” whether the underlying cause is central or peripheral. While communication is often best established by vertical eye movements following pontine stroke (fig 1), with other conditions such as severe neuropathy or extensive acute disseminated encephalomyelitis (ADEM) (fig 2), a whole range of minimal movements may be exploited to facilitate communication—forehead winking, eye blinks, mouth movement and head or limb twitches. Communication aids, such as letter or picture boards, may be needed and it must be remembered that many patients may find it easier to communicate in their native language at times of stress. Achieving adequate communication is crucial in relieving the extreme frustration experienced by both the patient and relatives.

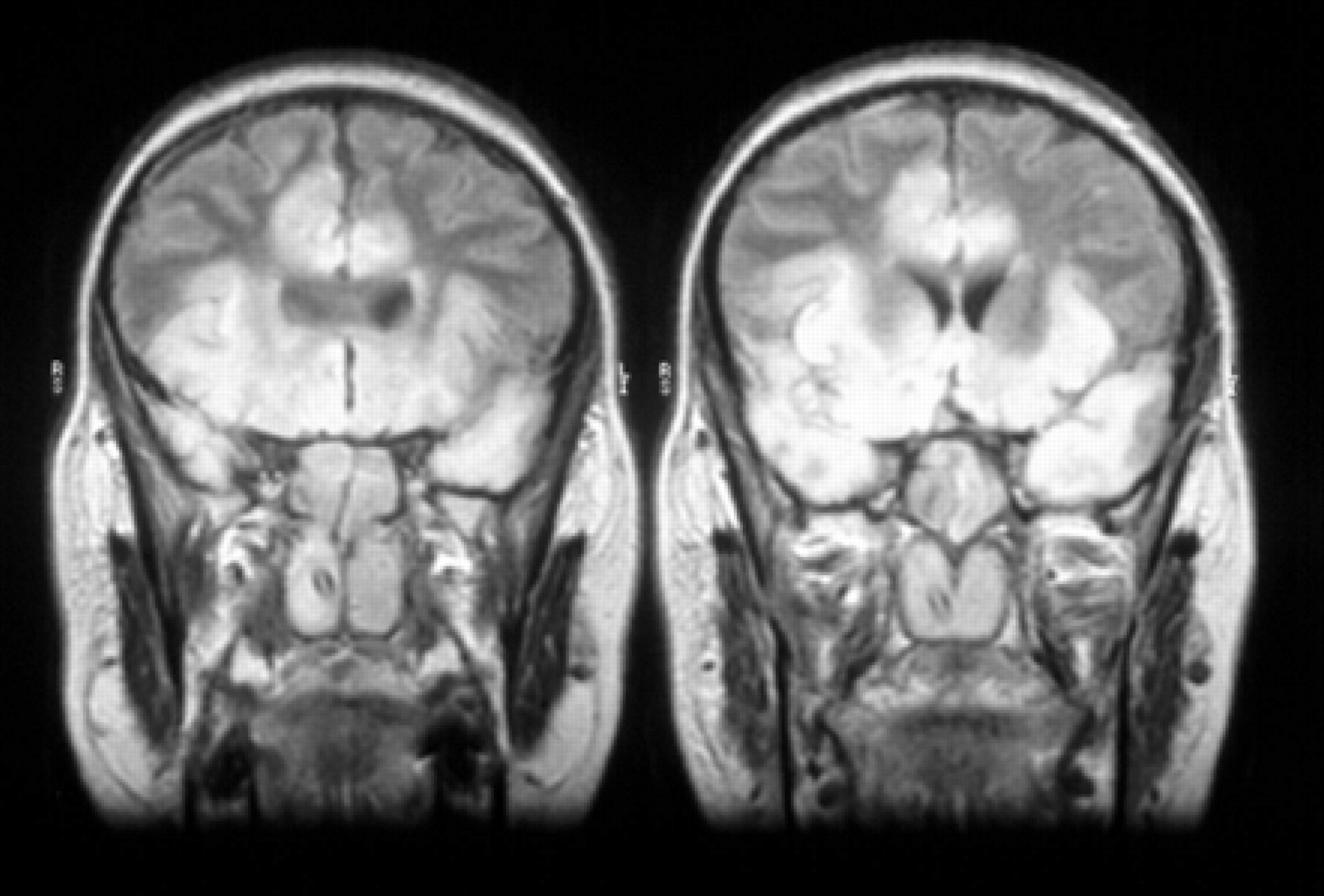
Even if effective communication cannot be established, the patient may still be awake and fully alert. The situation of being completely “locked in” is one of the most devastating and distressing clinical states, but is often overlooked. Although the EEG may help by showing awake and responsive rhythms, it is essential that the clinician recognises the possibility that their patient may be awake despite being unable to move.1, 2
The history
Following resuscitation it is crucial to obtain a clear history of the prodrome and presentation. This may be a difficult process requiring considerable patience, involving the patient, if possible, and also the relatives, observers, paramedics and A&E staff. This will include a history of:
Any previously known neurological disease, for example progressive disorders such as motor neuron disease (MND) and Duchenne muscular dystrophy, which may present acutely as a consequence of intercurrent events (usually aspiration pneumonia). Ongoing management decisions and any advanced directives made by the patient and their carers must be clarified quickly.
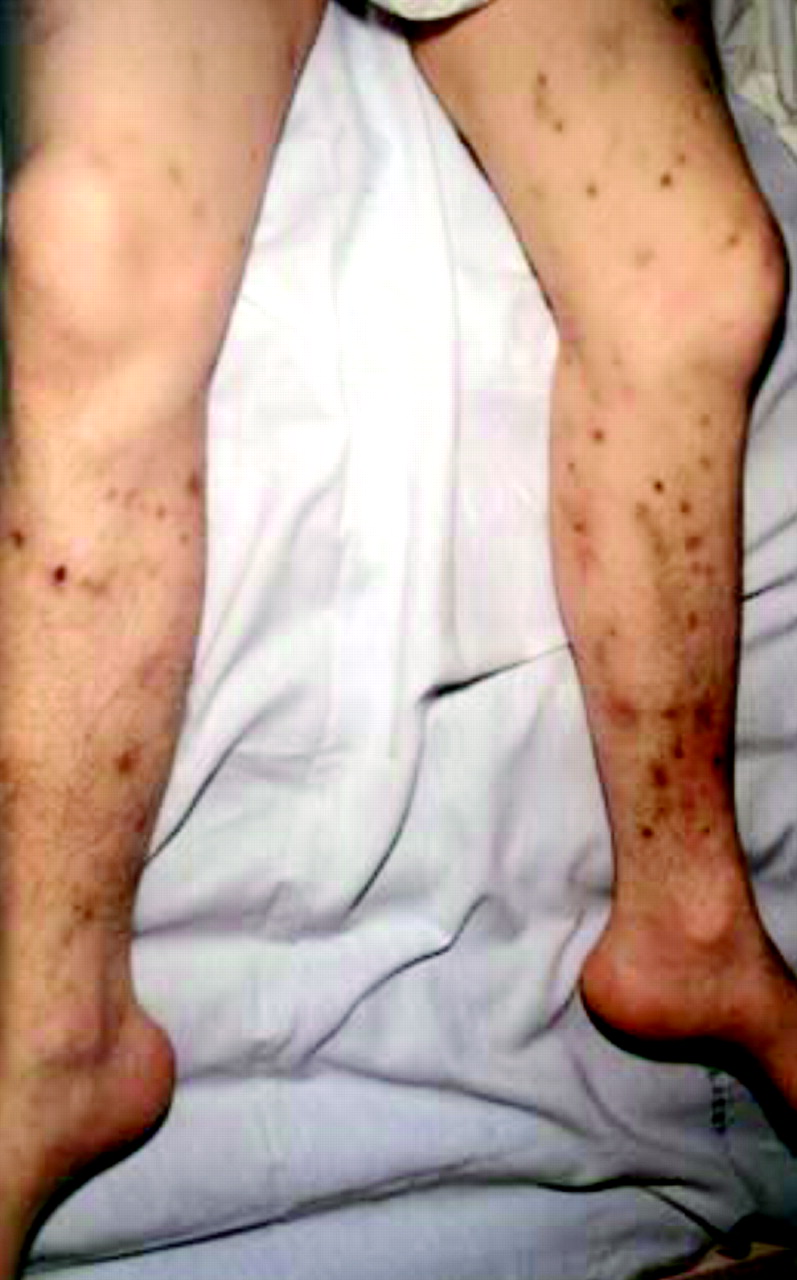
The prodrome, which may include symptoms of progressive morning headache, breathlessness and orthopnoea suggesting nocturnal hypoventilation due to respiratory muscle weakness (for example, myasthenia gravis, muscular dystrophy), preceding infective illness or diarrhoea (for example, Guillain Barré syndrome (GBS)), dietary factors such as tainted food that may have precipitated botulism, a history of drug ingestion or skin popping (fig 3) and any relevant family history.
The presence of systemic abnormalities, such as abdominal pain, may point towards porphyria or diabetes.
Progressive limb and trunk weakness developing over days or weeks suggests a progressive neuromuscular disorder, and a history of fatigability may indicate a neuromuscular junction abnormality.
The rate and pattern of onset is a guide to both diagnosis and prognosis—how did any bulbar, respiratory muscle, limb and trunk weakness develop? Was admission precipitated by an acute intercurrent event, was intubation undertaken acutely or on a semi-elective basis, and was a tracheostomy necessary? In GBS a worse prognosis is suggested by a rapid deterioration to ventilation and early, severe bulbar involvement in myasthenia gravis or MND may predispose to aspiration and sepsis.

The examination
Examination provides important clues to the diagnosis but is frequently difficult for the reasons described above:
Ophthalmoplegia might suggest GBS or a brainstem lesion, and an associated ptosis indicates myasthenia.
Poorly responsive pupils are a clue to botulism.
Bulbar function is extremely difficult to characterise at the bedside and poor tongue movement, palatal excursion and absent pharyngeal reflex do not necessarily imply that bulbar weakness is part of the underlying condition.
Neck flexion and shoulder abduction often give a helpful guide to coexisting weakness of the respiratory muscles and in particular the diaphragm.
In the limbs the presence of fasciculation and upper motor neuron signs may point towards MND whereas fatigable weakness indicates a neuromuscular junction abnormality and peripheral weakness with sensory loss suggests neuropathy.
In the poorly responsive patient, a central cause should be suspected if there are brisk reflexes; however, a pyramidal distribution of weakness may be misleading as this is often seen in peripheral neuropathy as well as upper motor neuron disorders.
Variability in the signs should also be recognised and recorded as acute flaccid weakness may rarely be functional.
The patient who has been on the ICU for a long time
In patients who are referred following a prolonged ICU admission a meticulous assessment of the preceding history and ICU care must be undertaken. It is all too common for patients to be referred with little background information about the underlying condition, intercurrent events, drug treatment and metabolic factors. It is particularly important to sort out:
Any systemic complications of the underlying disorder or ICU admission which may have occurred—in particular a history of hypoxic-ischaemic brain damage, sepsis, systemic inflammatory response syndrome (SIRS), organ failure, metabolic or endocrine abnormalities, or endocarditis.
Medication used during the ICU admission, especially sedation, anaesthesia, antibiotics, neuromuscular blocking agents and steroids.
Assessment of the adequacy of feeding and hydration.
The pattern of weakness: distal wasting, weakness and sensory impairment, suggests critical illness neuropathy, while proximal limb girdle and truncal weakness point towards critical illness myopathy. However, caution is necessary because an upper motor neuron pattern of weakness is commonly due to nerve or muscle disorders rather than a spinal cord lesion, and focal limb weakness is more often the consequence of nerve entrapment (for example, ulnar, femoral or common peroneal nerve palsies) than stroke.
The pattern of breathing on attempted weaning may also be an important guide. If there is a central brainstem lesion interfering with the generation of respiratory rhythm there will be no volitional respiratory movement; however with respiratory muscle weakness there may be a partial ventilatory response which is inadequate to maintain ventilation without support. With selective involvement of the phrenic nerve or diaphragm weakness there may be prominent orthopnoea and paradoxical movement of the diaphragm, which can be observed in patients who are able to breathe spontaneously; inward movement of the abdomen on inspiration when the patient is supine, particularly during a sniff manoeuvre, because the paralysed diaphragm is unable to contract on inspiration and therefore passively ascends with the chest wall (the paralysed diaphragm descends on inspiration when the patient is erect because of gravity and therefore diaphragm excursion appears normal).
Seizures may be difficult to recognise in the paralysed patient and the only clue may be isolated, recurrent twitching of small muscle groups in the face or limbs. On the other hand, a prolonged post-ictal Todd’s paresis may appear as unexplained focal muscle weakness.
Intercurrent events which may have occurred in the ICU include stroke, acute disseminated encephalomyelitis, and central pontine myelinolysis. Furthermore it is important to recognise that GBS may develop during the course of an acute illness in a patient admitted to ICU for other reasons.
Occasionally the presence of systemic features, particularly pneumonia, sepsis and multi-organ failure, may obscure a neurological cause for the primary presentation (for example, GBS or MND). Only when these have been adequately treated is the underlying diagnosis recognised.
Persistent weakness or failure to wean following a routine anaesthetic may suggest either an intercurrent event has occurred (for example, stroke) or that a previously unsuspected neuromuscular condition has become symptomatic (for example, congenital myopathies and myasthenia, acid maltase deficiency, myotonic dystrophy or MND). In patients who fail to wean following cardiac surgery, impaired diaphragm function manifest as orthopnoea or failure to wean when supine may suggest phrenic nerve damage, while focal distal weakness and cortical sensory loss may indicate a boundary zone ischaemic stroke.
Investigations
Routine investigations include haematology, electrolytes, thyroid function, creatine kinase and autoantibody screen.
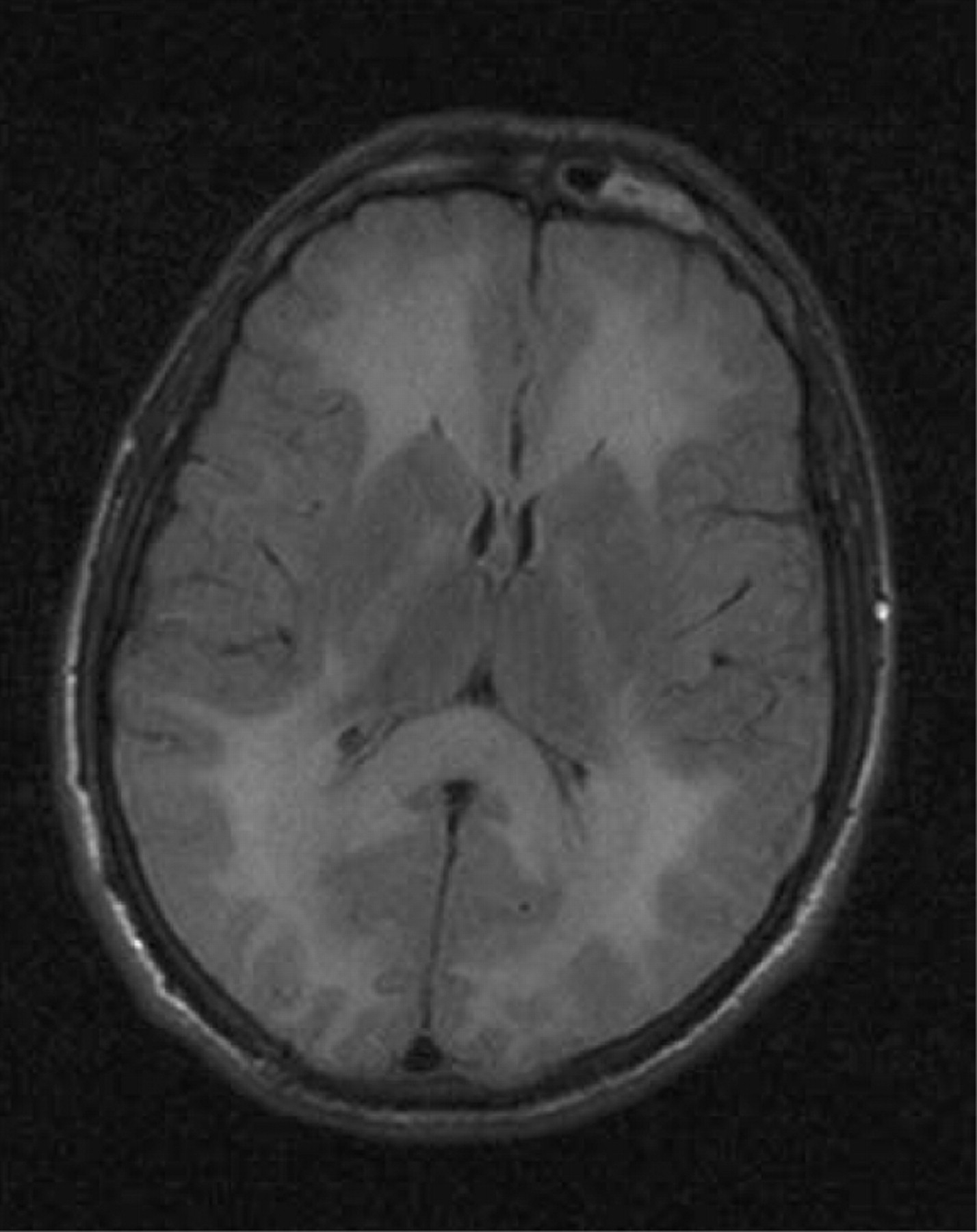
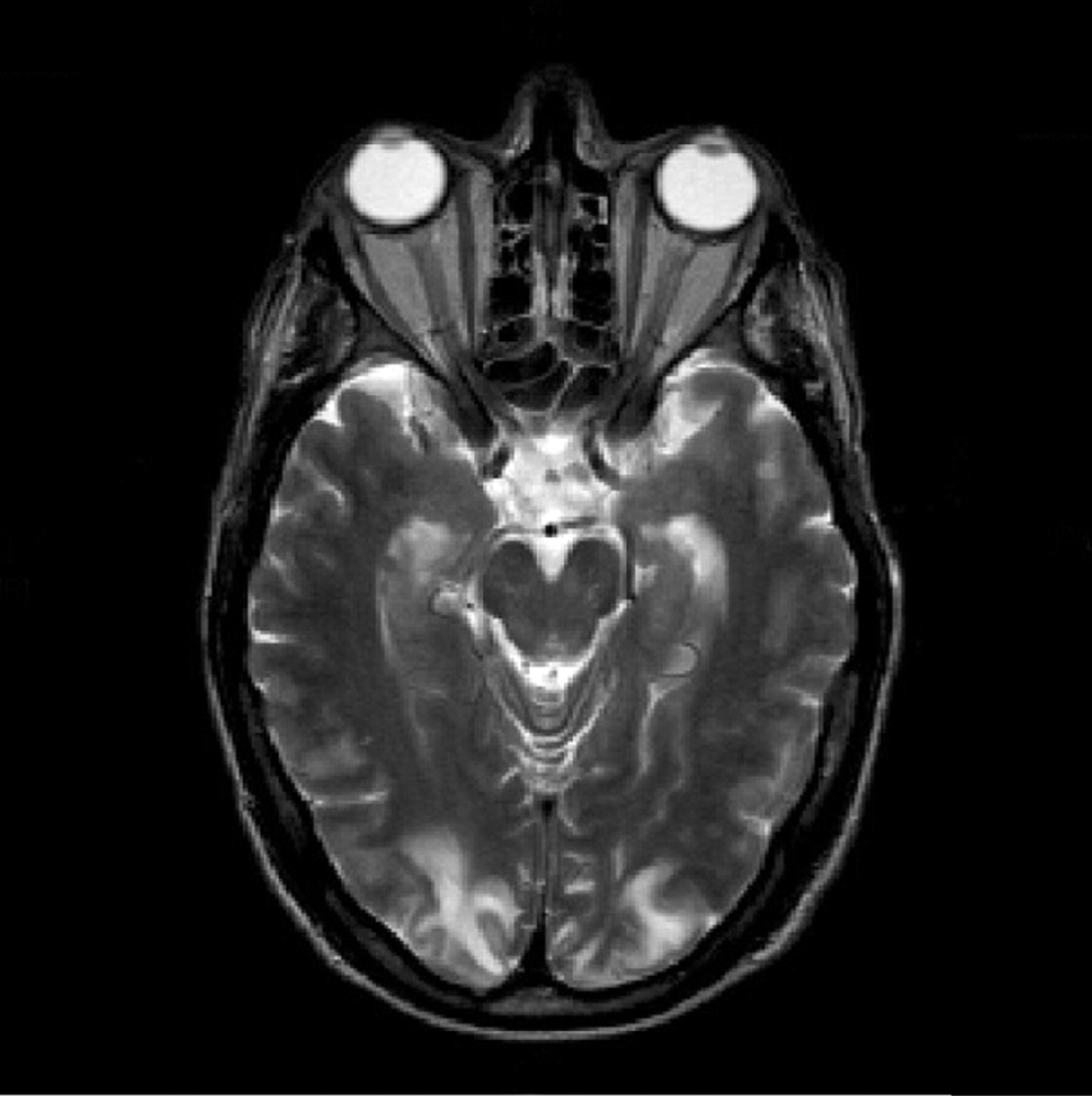
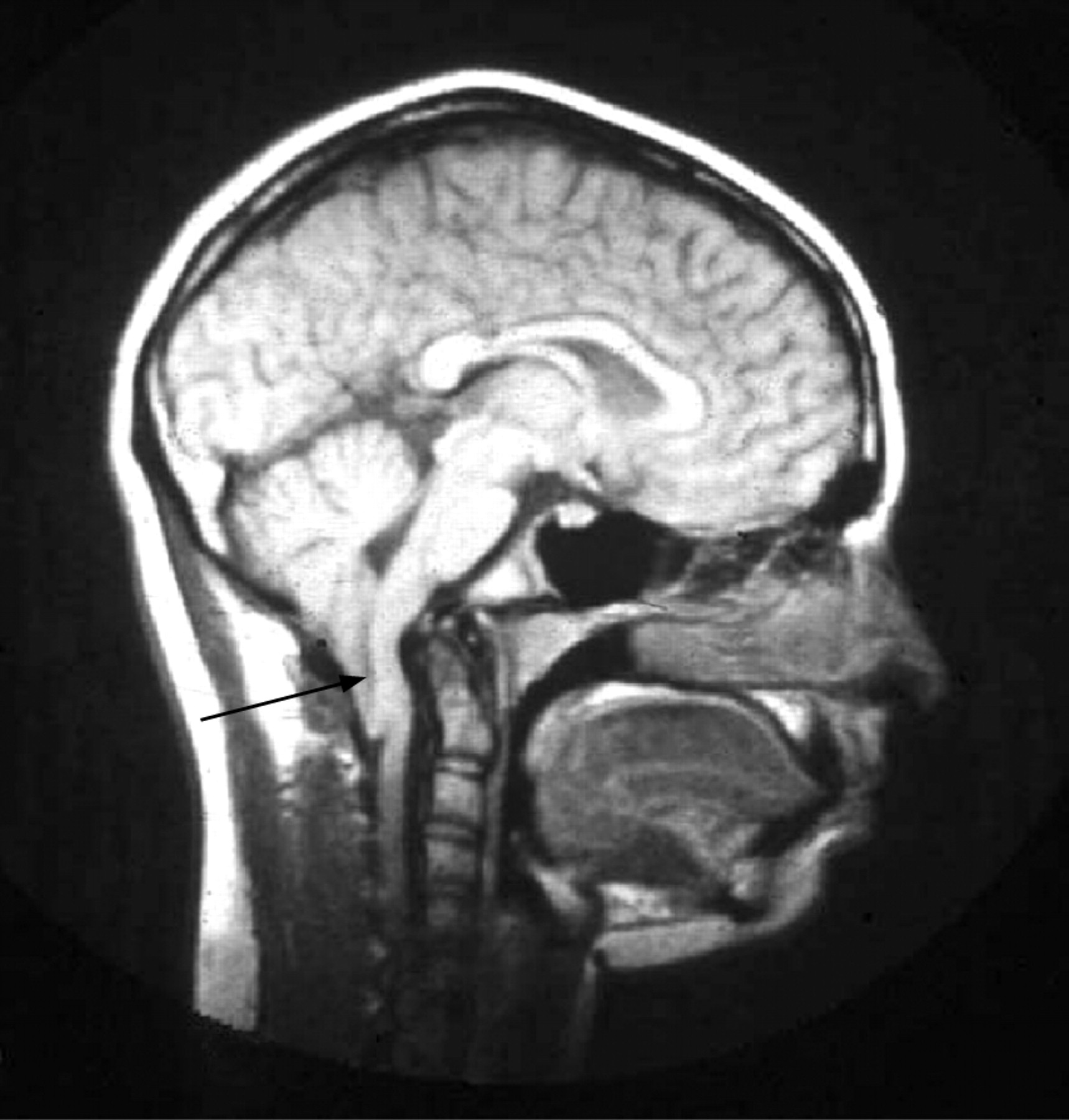
Imaging may show a central disorder such as stroke, ADEM, toxic encephalopathy (fig 4), central pontine myelinoysis (fig 5) or posterior reversible leukoencephalopathy (PRES) (fig 6).
An EEG may be valuable in showing partial epilepsy and also response awake rhythms in a “locked in” patient.
Specific bioassays may be necessary if botulism is suspected.
Cerebrospinal fluid (CSF) examination may show albumine-cytological dissociation or malignant infiltration.
Neurophysiology lies at the core of the differential diagnosis even though EMG on the intensive care unit may be difficult because of lack of cooperation from the patient, poor electrical screening, and technical difficulties because of the presence of other electrical equipment and non-availability of adequate portable machinery. In some units there is lack of an available neurophysiologist and indeed neurophysiology may simply be unavailable. In this situation it is unsatisfactory for a patient to continue with unexplained ventilator dependence or weakness. As soon as it is safe, if local facilities are not available, such patients should be transferred to an appropriate specialist unit to establish the diagnosis and get advice on further management.
Occasionally nerve biopsy may be necessary if nerve conduction shows severe axonal loss but a primary demyelinating or vasculitic aetiology is suspected, and sometimes even a muscle biopsy.


In summary, following initial clinical assessment of the patient who is not receiving sedation or neuromuscular blocking agents, it is necessary to establish whether the weakness is due to the causes listed in table 1. However, it is important to remember that weakness is often multifactorial and any combination of these factors may be relevant in an individual patient.
FACTORS ASSOCIATED WITH WEAKNESS FOLLOWING CRITICAL ILLNESS
See table 2 for details.
Sepsis
Systemic inflammatory response syndrome (SIRS) is a severe systemic response that occurs in up to 50% of those in a critical care setting due to infection or other insults such as burns, trauma or surgery (table 3). It is associated with organ dysfuction, hypotension, hypoperfusion, lactic acidosis, oliguria and an altered mental state.
Septic encephalopathy is the commonest form of encephalopathy encountered in intensive care medicine and is present in 50–70% of septic patients. Although the pathogenesis is not clearly understood it is thought to be related to bacterial toxin release, high fever and the effects of mediators of inflammation. It is characterised by obtundation, disorientation, delirium, impaired consciousness or coma. There may be rigidity, tremors and seizures. The outcome depends on the precipitating condition and coexisting renal and hepatic impairment but it is potentially reversible; the aims of treatment are removal of the underlying source of sepsis and supportive intensive care.
Sedation and analgesia
These are confounding factors which may cause depressed consciousness and weakness. Withdrawal of sedation before review is essential for reliable neurological assessment and it is important to realise that the effects of sedative drugs may take several days to clear; any of them may accumulate if there is multi-organ failure, and even with apparently normal metabolic function their half-life may be abnormally long in patients who have been critically ill. Deep sedation leads to prolonged recovery time, difficulty weaning, ileus and a confusional state. Other drugs used for sedation or analgesia may also contribute to apparent neuromuscular weakness; opiods (particularly morphine, fentanyl, alfentanyl), benzodiazepines and other anxiolytic and hypnotic agents.
Anaesthetics
Propofol is now widely used as an anaesthetic agent because it rapidly crosses the blood-brain barrier, produces a marked hypnotic effect, does not undergo renal or hepatic metabolism and is rapidly reversible. However, awakening may be significantly prolonged with high infusion doses over many days and there is no antagonist or reversing agent available.
Midazolam is a short acting benzodiazepine widely used because it causes amnesia, is easily reversed by flumazanil and is well tolerated. However, prolonged sedation may occur after discontinuing the drug in patients with renal or hepatic failure.
Neuromuscular blocking agents, particularly with prolonged use in the presence of other agents which may affect neuromuscular junction function, may lead to prolonged neuromuscular blockade or critical illness polyneuropathy or myopathy (see below) (table 4).
CAUSES OF WEAKNESS ON THE ICU
See table 5 for details.
Brain disorders
These may present with respiratory failure, occasionally with acute flaccid weakness although, in most cases, consciousness is impaired. Brainstem stroke presents acutely, but extensive brainstem involvement can also occur with severe infectious encephalopathy (brainstem encephalitis or herpes simplex encephalitis) or extensive demyelination (ADEM, central pontine myelinolysis) and lead to severe weakness in the cranial nerves and limbs and a complete inability to initiate any spontaneous respiratory movements.
Spinal cord
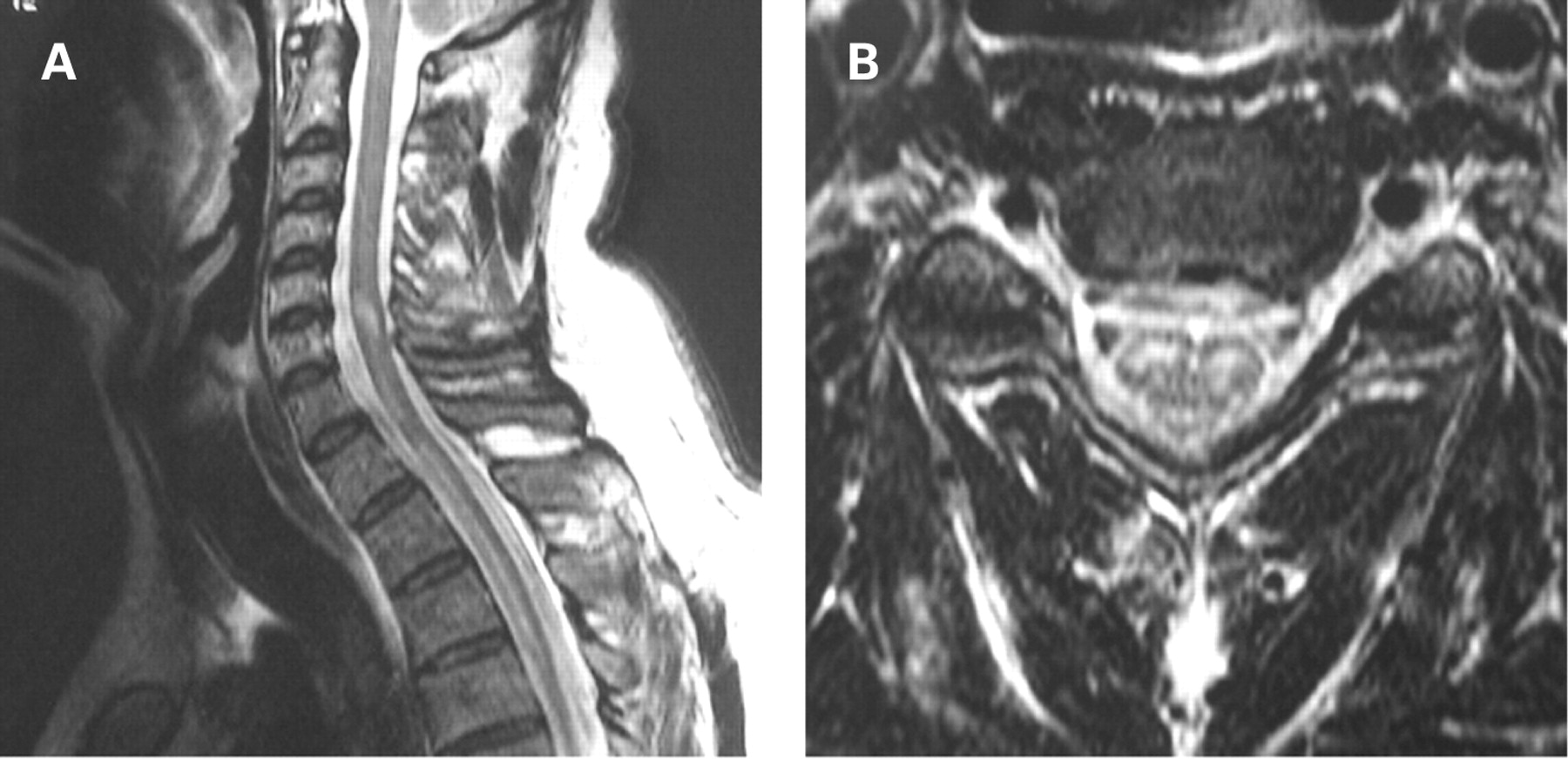
Traumatic, demyelinating or vascular lesions of the spinal cord (fig 7), particularly at high cervical levels, may cause acute flaccid tetraplegia, selectively affecting respiratory control. Patients with previously unsuspected Arnold Chiari malformation with cerebellar ectopia (fig 8) may even present with acute ventilatory failure and tetraparesis following a cervical spine hyperextension injury. Cervical myelitis (fig 9) or structural lesions of the cord may selectively affect respiratory control, in particular the automatic generation of the respiratory rhythm leading to central apnoea. Of course surgery to the high cervical cord may also lead to respiratory paralysis.


Anterior horn cell
Motor neuron disease may present with acute respiratory weakness, or patients may develop this early in the course of their disease—often associated with infection. These patients may be intubated and ventilated before the diagnosis has been made. The progressive nature of the condition means that weaning to non-invasive ventilatory support may be impossible.3, 4
Patients with late post-polio functional deterioration may also present with progressive nocturnal hypoventilation or acute respiratory failure. And acute flaccid paralysis may be due to acute poliomyelitis or West Nile Virus, or occur as a paraneoplastic syndrome.
Neuropathies
Patients can be admitted because of a severe, primary neuropathy which recovers slowly (for example, acute inflammatory demyelinating neuropathy), or develop neuropathy related to therapeutic agents (for example, acute intermittent porphyria) or nutritional deficiencies. And the intensive care patient is at particular risk of developing critical illness polyneuropathy as a complication of prolonged treatment.
Acute inflammatory demyelinating polyneuropathy. About one third of GBS patients require admission to ICU because of respiratory insufficiency requiring mechanical ventilation, severe bulbar weakness threatening pulmonary aspiration, or autonomic instability causing cardiac arrhythmias or fluctuations in blood pressure. Ventilatory failure is primarily caused by inspiratory muscle weakness, although weakness of the abdominal and accessory muscles of respiration, retained airway secretions leading to pulmonary aspiration and atelectasis all contribute. Any associated bulbar weakness and autonomic instability reinforce the need for control of the airway and ventilation.5, 6
Raised CSF protein levels accompanied by no or only a few mononuclear cells occurs in over 80% of GBS patients after two weeks. If there is a pleocytosis of more than 10 lymphocytes/mm3, neuropathies related to Lyme disease, recent HIV infection, or sarcoidosis must be excluded by appropriate investigation.
An acute demyelinating polyneuropathy can rarely complicate the post-transplantation course in allograft recipients, more frequently bone marrow than solid organ transplantation. It may also follow cytomegalovirus infection in immunocompromised patients, or as an HIV seroconversion illness.
The diagnosis of neuropathy may be difficult because of the presence of multi-organ impairment or other general medical factors. Occasionally, some patients may actually develop acute demyelinating polyneuropathy during the course of a critical illness; not surprisingly when this occurs, the rapid development of inexplicable weakness and its cause may not be recognised.
Miller Fisher syndrome. Ataxia, areflexia and ophthalmoplegia are the classical features.7–9 Diplopia is the most common initial complaint (in about one third), while ataxia is evident in about one fifth of patients at onset. Facial weakness, bulbar impairments with dysphagia and dysarthria, along with facial and limb paraesthesias may also occur and mild proximal limb weakness is found in approximately one third of patients.
Bickerstaff’s brainstem encephalitis is a closely-related condition in which progressive ophthalmoplegia and ataxia, maximal at less than 4 weeks, are associated with drowsiness and coma, and upper motor neuron signs. MRI shows high intensity abnormalities in the posterior fossa, white matter or thalami in 30% of patients. The course is monophasic with complete remission at 6 months in two thirds.8, 9
Acute motor and sensory axonal neuropathy (AMSAN) and acute motor neuropathy (AMAN) are axonal neuropathies which may present with paralysis developing rapidly, over hours, leading to respiratory failure.10 Indeed, there may be severe, extensive and occasionally total paralysis of all voluntary muscles of the body, including the cranial and ocular muscles. CSF protein is raised but there should be <10 white blood cells/mm3. In AMAN and AMSAN all peripheral nerves, including cranial nerves, may be unresponsive to electrical stimulation, even when the stimuli are of high voltage and long duration. Needle EMG shows abundant fibrillation potentials after three weeks.
Acute intermittent porphyria is an uncommon autosomal dominant disease, characterised by recurrent episodes of abdominal pain, psychiatric disturbances, seizures and acute, predominantly motor axonal, neuropathy with autonomic features, which may mimic GBS causing bulbar and ventilatory failure with patchy sensory involvement. Attacks can be precipitated by heavy alcohol consumption and by numerous medications, many of which are commonly administered in the ICU, such as diazepam, theophylline and barbiturates.
Phrenic nerve neuropathies may lead to ventilatory failure and admission to ICU (table 6).
Critical illness polyneuropathy is an acute sensorimotor axonal neuropathy which develops in the setting of SIRS, septic encephalopathy and/or multi-organ failure—particularly in the presence of hypoalbuminaemia, hyperglycaemia and insulin deficiency, with or without corticosteroids and neuromuscular blocking agents.11–15 It is characterised by delayed weaning, severe distal flaccid wasting and weakness, areflexia and sensory impairment in those patients who are able to cooperate with the examination. A pure motor neuropathy in association with neuromuscular blocking agents is a variant which has been reported.16–21 The signs are difficult to elicit because of the difficulties in examining an uncooperative patient who is sedated or has coexistent septic encephalopathy. Moreover, it is often difficult to record and interpret neurophysiology studies on the ICU, particularly if there is lower limb oedema. And to compound the diagnostic problem, neuropathy, neuromuscular blockade and myopathy may all coexist in the same patient.13 Nerve biopsy and autopsy studies have shown axonal degeneration of both sensory and motor fibres without evidence of significant inflammation or primary demyelination. Muscle shows scattered atrophic fibres in acute denervation and grouped atrophy in chronic denervation.22
The differential diagnosis includes other neuropathies, neuromuscular transmission defects and myopathies, of which the most important is critical illness myopathy (see below). The diagnosis of neuromuscular blockade can be established by repetitive nerve stimulation studies. Differentiating critical illness polyneuropathy from critical illness myopathy is more difficult and sometimes these two disorders occur simultaneously. As patients are often sedated, quantitative electromyography is not possible. Stimulation of muscle directly and indirectly (by stimulating the nerves to the muscle) may provide some more information but a reliable diagnosis can only be achieved after muscle biopsy.23–27
Critical illness polyneuropathy is self-limiting and the prognosis is influenced by the severity of the underlying condition which itself accounts for most of the mortality. The outcome is also related to the severity of the sepsis and other factors, including the extent and severity of the neuropathy, the time on ICU, and the presence of hyperglycaemia, hyperosmolality or hypoalbuminaemia.28–31 When the neuropathy is mild or moderate recovery is relatively rapid and complete. In those with more severe polyneuropathy recovery is limited; persisting weakness and sensory deficits are common in long-term survivors of protracted critical illness, even up to four years after discharge.
Neuromuscular junction disorders
Disruption of neuromuscular transmission is an important cause of weakness and failure to wean in critically ill patients.
Neuromuscular junction (NMJ) blocking agents are used increasingly to facilitate intubation, improve lung compliance, allow more efficient mechanical ventilation and reduce fluctuations in raised intracranial pressure. Drugs that potentiate the depth of motor blockade may also prolong recovery (table 4). NMJ blocking agents may be divided into depolarising (suxamethonium) and non-depolarising (for example, vecuronium, pancuronium, atracurium):
Depolarising agents physically resemble acetylcholine and, therefore, bind to, activate and block acetylcholine receptors. Suxamethonium is short acting (2–5 min) and produces intense neuromuscular relaxation by causing prolonged depolarisation of the postsynaptic receptors at the NMJ. Its sole use is to facilitate tracheal intubation. It should be avoided in neuromuscular disease as hyperkalaemia may follow its use and risk the development of malignant hyperthermia.
Non-depolarising agents bind reversibly to postsynaptic acetylcholine receptors, antagonising acetylcholine, but do not activate them. They produce longer lasting neuromuscular blockade and doses are cumulative, particularly if there is renal or hepatic failure. The effects are enhanced by hyperkalaemia, hypophosphataemia and hypermagnesaemia.
Prolonged neuromuscular blockade is defined as recovery from NMJ blocking agents 50–100% longer than predicted by pharmacological parameters. It is particularly associated with steroid-based NMJ blocking agents and occurs after either short- or long-term blockade with non-depolarising agents (vecuronium is the most commonly reported). It may be associated with prolonged use or high doses of these drugs, metabolic acidosis, hepatic or renal insufficiency, hypermagnesaemia or in association with corticosteroids, aminoglycosides or other anaesthetic agents. Weakness should not persist beyond two weeks after stopping the blocking agent and typically lasts for only a few days. Prolonged blockade should be considered in any patient who remains weak after discontinuation of NMJ blocking agents.32
Repetitive nerve stimulation is abnormal with a decremental response in the compound muscle potential amplitude but the blood creatine kinase (CK) levels are normal. “Train of four” stimulation, in which four equal pulses are delivered over 2 seconds, is used to assess recovery from acute block. Lack of an attenuated response indicates prolonged neuromuscular blockade but formal repetitive nerve stimulation is required for confirmation.
Neuromuscular disease can be unmasked in previously undiagnosed cases by medications commonly used intraoperatively and in the recovery room or ICU. Patients with myasthenia gravis or the Lambert-Eaton myasthenic syndrome (LEMS) have an extremely high sensitivity to subtherapeutic levels of agents that have minimal effect on neuromuscular transmission in normal people (table 4).
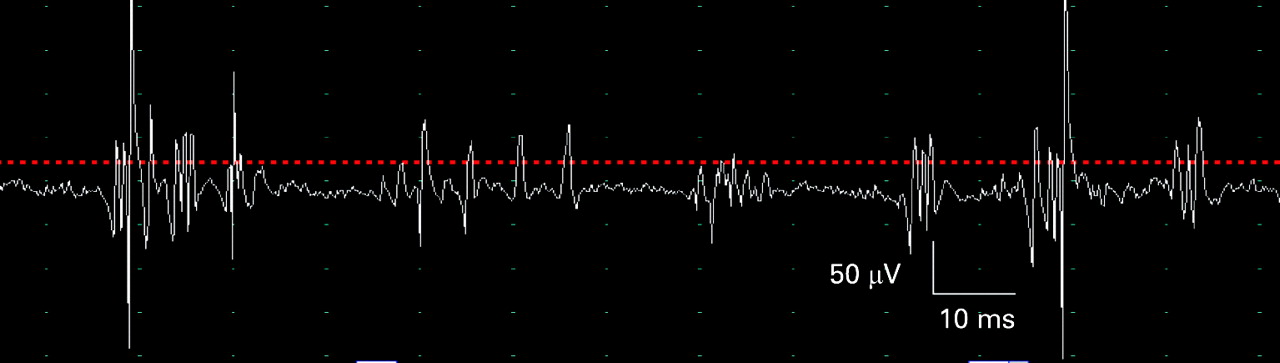
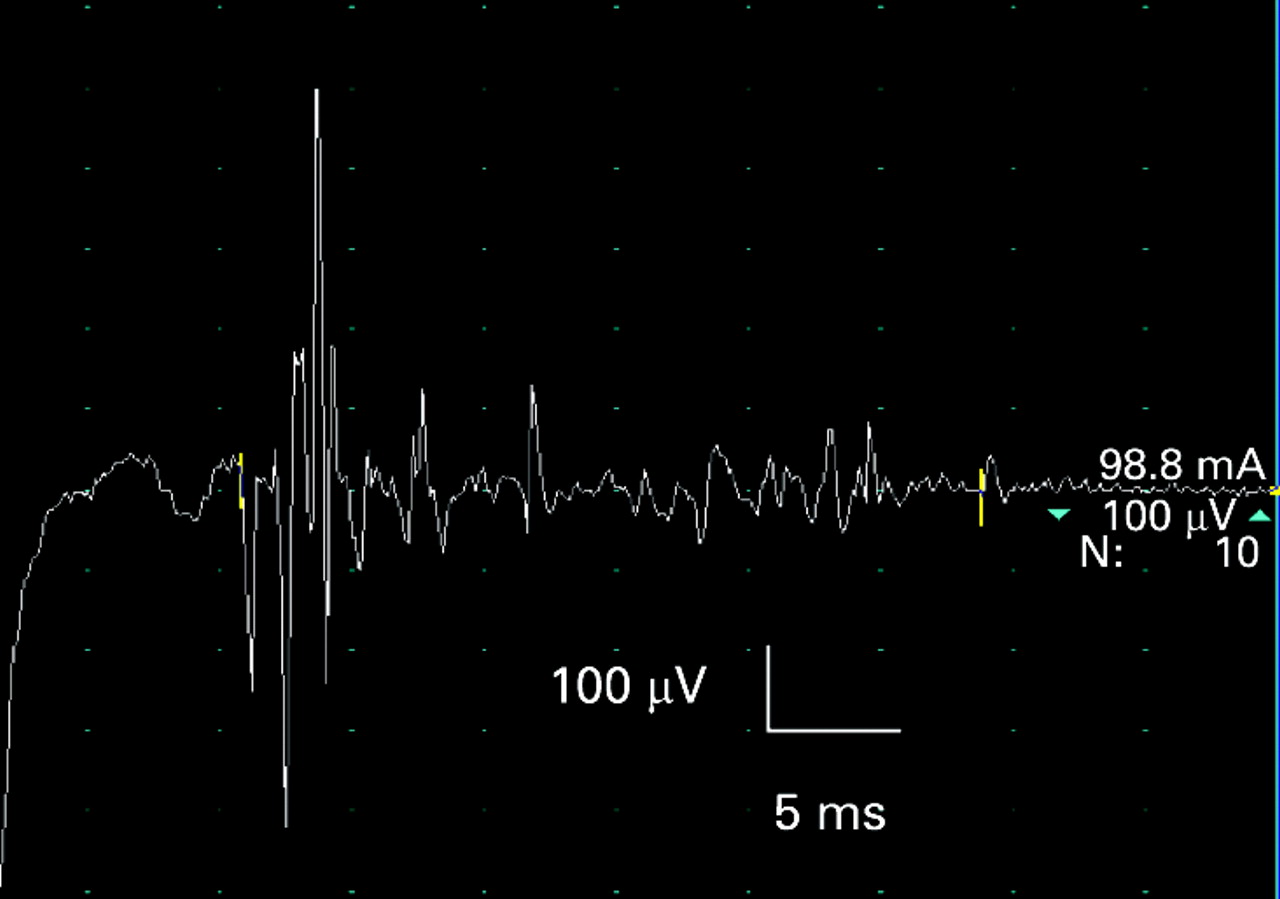
Myasthenia gravis. Some patients present with respiratory failure due to a myasthenic crisis (usually precipitated by infection, surgery or inadequate treatment) and others may develop respiratory failure during the course of their disease, sometimes caused by a therapeutic cholinergic crisis (if the patient requires ventilatory support it is not necessary to distinguish between myasthenic and cholinergic crisis at presentation).33–36 The associated bulbar weakness predisposes to pulmonary aspiration and acute respiratory failure necessitating urgent tracheal intubation and ventilation. Repetitive stimulation typically shows a decrement which is maximal after the 4th or 5th stimulus, and which is more marked at 3 Hz stimulation than at slow rates, consistent with a post-synaptic disorder or neuromuscular transmission. There is post-activation repair of decrement immediately after exercise, and post-activation exhaustion when retested after 3 minutes of rest (fig 10A–D). Single-fibre EMG shows increased jitter (fig 10E) and blocking in weak muscles.

Botulism is caused by toxins produced by Clostridium botulinum, an anaerobic gram positive organism. These act at the presynaptic region of the NMJ causing failure of release of acetylcholine. The classical form occurs after ingestion of food that contains toxin but increasing numbers of patients are being seen as a consequence of using contaminated opiates or unclean needles to inject drugs of abuse into infected skin lesions (skin popping, fig 3). Patients develop cranial nerve deficits, including blurred vision, diplopia, ptosis, dysarthria, and dysphagia. The weakness often affects the arms before progressing to the legs. Autonomic symptoms, including dry mouth, unreactive pupils, and ileus, are frequent. The condition may be suspected by prominent and early ocular signs (dilated sluggishly reactive pupils and ophthalmoplegia) and dysphagia.
Definitive diagnosis requires detection of toxin in the patient’s serum, stool, or food by bio-assay but this takes too long. Unfortunately, electrodiagnosis may not be straightforward because the findings depend on the timing of examination and the severity of the disease. Patients with relatively mild disease may have “normal” or low-normal compound muscle action potential (CMAP) amplitudes, show no clear decrement at low rates of repetitive stimulation, but demonstrate an increment of >40% after exercise or after high rates of repetitive stimulation, consistent with this being a presynaptic disorder of neuromuscular transmission. However, this increment is usually not as marked as in LEMS and is generally only seen in clinically weak muscles. Unlike LEMS and myasthenia gravis, there is no post-activation exhaustion and the increment usually persists for 4–20 minutes. In more severe disease, the resting CMAP is usually small, there may or may not be decrement at low rates of repetitive stimulation, and the incremental response to high rates of stimulation may be minimal or absent.37 Needle EMG may show fibrillation potentials and positive sharp waves in severely affected muscles, and small amplitude potentials resembling a myopathic pattern. Both amplitude and interference pattern may increase with sustained effort. Single-fibre EMG is often abnormal early in the disease, with increased jitter and blocking in affected muscles. As in LEMS, the amount of jitter decreases with an increase in the muscle fibre’s firing rate.
Other neuromuscular transmission disorders which may cause confusion in ICU include organophosphate poisoning, tick paralysis, black widow spider and certain types of snake envenomation.
Muscle disease
According to neurophysiological38 and biopsy39 studies, myopathies occur much more frequently during critical illness than previously recognised. Three main types have been identified: diffuse non-necrotising cachectic myopathy, myopathy with selective loss of thick (myosin) filaments (“critical illness myopathy”) and the acute necrotising myopathy of intensive care.40
Diffuse non-necrotising cachectic myopathy is common and presents as muscle wasting with associated weakness. However the CK levels and EMG are normal or show only mild changes. It is associated with prolonged ICU admission, sedation or paralysis causing muscle disuse, and poor nutrition with protein catabolism during critical illness. There is proximal or general weakness. Biopsy shows type 2 fibre atrophy and neurogenic atrophy.
Critical illness myopathy is a distinct form of myopathy which occurs in patients with critical illness on ICU (other names have included myopathy with selective loss of thick filaments, acute quadriplegic myopathy, acute illness myopathy, acute myopathy of intensive care, rapidly evolving myopathy with myosin-deficient fibres). It is under-recognised and is one of the most common forms of generalised weakness on the ICU, being considerably more frequent than critical illness polyneuropathy.41, 42 It is sometimes associated with prolonged exposure to high doses of corticosteroids and non-depolarising muscle blocking agents used to treat acute pulmonary disorders, such as asthma. However, it can occur in other situations including systemic inflammatory response syndrome (SIRS), major organ transplantation, particularly liver43, 44 and it does not seem to correlate with the duration of intensive care.45 Other factors that may contribute include nutritional deficiencies, concurrent drug administration with aminoglycosides or cyclosporin, hyperglycaemia, renal and hepatic dysfunction, fever, severe metabolic and electrolyte disorders.
The problem tends to be recognised as the acute illness resolves when it becomes apparent that the patient cannot wean from ventilatory support because of limb and respiratory muscle weakness. The limb weakness may be mild or severe and is predominantly proximal although it may be generalised. There may be facial and neck weakness as well, but the extraocular movements are often spared, reflexes are reduced and sensation is not affected.
Blood CK levels are raised in <50% and electrodiagnostic studies are neither sensitive nor specific for diagnosing myopathy in the critically ill. Nevertheless, a few diagnostically helpful features have been noted:
CMAP durations are often prolonged46–48 (fig 11) with a reduction in the mean muscle fibre conduction velocities, as well as a wider spread of velocities due to the presence of very slow conducting fibres49–51 (fig 12).
Needle EMG may show myopathic features (fig 13) but is often unhelpful in distinguishing between myopathy and polyneuropathy because insufficient motor units are obtained. Fibrillation potentials and positive sharp waves can be present in critical illness myopathy and polyneuropathy.


Firm diagnosis needs the invasive procedure of muscle biopsy, and hence cannot be achieved with ease. Histological changes include abnormal variation of muscle fibre size, fibre atrophy, angulated fibres, internalised nuclei, rimmed vacuoles, fatty degeneration of muscle fibres, fibrosis, and single fibre necrosis.39 There are no inflammatory changes (fig 14). There is selective loss of thick (myosin) myofilaments and preservation of thin (actin) myofilaments and Z discs (fig 15).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The outcome seems to be much better than after critical illness polyneuropathy and most patients make a full recovery unless there has been severe and prolonged paralysis.
Acute necrotising myopathy of intensive care is a rare condition which may be a form of rhabdomyolysis. It develops after exposure to neuromuscular blocking agents with or without steroid therapy, but may be associated with other infective or metabolic insults. The serum CK is markedly elevated, there is usually associated myoglobinuria, EMG confirms a severe myopathy and biopsy shows patchy or widespread necrosis with occasionally vasculitis or infarction within the muscle. The prognosis for recovery of weakness is poor.
Other myopathies. Almost any form of myopathy may be responsible for prolonged respiratory failure in critical care units:52, 53
Patients with weakness and asthma and others who have been exposed to corticosteroids alone may have acute steroid myopathy. This is a slowly-evolving, mild-to-moderate proximal weakness with mild elevation of CK and type 2 fibre atrophy.
Occasionally, direct sepsis may affect the muscle, causing pyomyositis due to septic micrometastases.
Inflammatory myopathies cause respiratory muscle weakness, and patients with dermatomyositis may have a characteristic skin rash. The EMG may be misleading, showing numerous fibrillation potentials and positive sharp waves that are more characteristic of denervation from neuropathy. The CK is raised and muscle biopsy of an involved muscle is necessary to make the diagnosis. Other inflammatory myopathies include viral myositis, acute alcoholic rhabdomyolysis and myopathy secondary to hypophosphataemia. Trichinosis caused by the ingestion of raw contaminated pork may cause an acute and severe myopathy with diaphragm and intercostal muscle involvement.
Patients with muscular dystrophy may present with ventilatory failure or develop respiratory insufficiency either as an early manifestation or as an inevitable feature of disease progression. Myotonic dystrophy is occasionally identified for the first time in the ICU. In adults, acid maltase deficiency may present with proximal weakness, scoliosis and diaphragmatic paralysis; the apparently abrupt respiratory insufficiency probably occurs on the background of longstanding unrecognised disease. Progressive respiratory impairment occurs in all patients with Duchenne muscular dystrophy, and acute deterioration may occur due to intercurrent events such as aspiration pneumonia.54
Rhabdomyolysis may be precipitated by trauma, compartment syndrome, ischaemic arterial occlusion or drugs and is associated with high serum CK levels, myoglobinuria and general weakness.
There are reports of respiratory decompensation associated with mitochondrial myopathies, limb girdle muscular dystrophy, HIV related myopathy and sarcoid myopathy involving the diaphragm.
Other conditions
Tetanus. The neurological manifestations are caused by tetanospasmin, a toxin elaborated in contaminated wounds by the gram-positive, spore-forming bacilli Clostridium tetani in unimmunised individuals.55 The toxin is transported via retrograde axonal transport into the spinal cord or brainstem, or both. It ultimately migrates to the presynaptic terminals and inhibits release of gamma-aminobutyric acid (GABA) and glycine, important inhibitory neurotransmitters. Most patients will be admitted to ICU because of increased muscle tone and spasms. Respiratory compromise is caused by spasm of respiratory muscles or laryngospasm. Autonomic dysfunction occurs in severe cases and results in heart rate and blood pressure lability, arrhythmias, fever, profuse sweating, peripheral vasoconstriction, and ileus. Muscle rupture and rhabdomyolysis can complicate extreme cases.
Paralytic rabies produces neuromuscular weakness that can be difficult to differentiate from other causes of weakness such as GBS.56 It may begin with local wound pain and paraesthesias followed by fasciculations near the site of inoculation.
PRACTICE POINTS
Neuromuscular disorders are important causes of failure to wean and prolonged morbidity in the intensive care unit (ICU).
Inability to communicate may be because the patient is “locked in” as a consequence of severe generalised paralysis.
The disorders may have been present in patients before admission to the ICU, or develop as a complication of their stay.
It is possible that weakness is due to an intercurrent event that may have occurred during the ICU stay.
Failure to wean from ventilatory support may also be due to isolated phrenic nerve palsy.
Critical illness myopathy and critical illness polyneuropathy account for much of this weakness, but other relatively common disorders, such as Guillain-Barré syndrome or myasthenia gravis, should be considered.
Careful review of recent medical history, neurological examination, and electrodiagnostic studies usually allow a definite diagnosis to be made; neurophysiology is crucial in distinguishing the different causes of weakness on the ICU.
Specific treatments may be available, but supportive care is the mainstay of management for most of the disorders.
The outcome is highly variable and is related to the severity of the underlying condition.
Acknowledgments
This article was reviewed by Richard Davenport, Edinburgh, UK.
REFERENCES
Other content recommended for you
- Weakness in the intensive care unit
- New weakness in a critically ill patient
- Approach to critical illness polyneuropathy and myopathy
- Neuromuscular disease and respiratory failure
- Mimics and chameleons in Guillain–Barré and Miller Fisher syndromes
- Weak at the knees
- Muscle diseases: mimics and chameleons
- Origin of ICU acquired paresis determined by direct muscle stimulation
- Admission to neurological intensive care: who, when, and why?
- Critical illness myopathy is frequent: accompanying neuropathy protracts ICU discharge