Article Text

Abstract
The diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is usually straightforward, but atypical presentations can represent a significant diagnostic challenge. This review highlights the clinical and electrophysiological ‘red flags’ that should make one consider an alternative diagnosis.
- NEUROPATHY
- NEUROPHYSIOL, CLINICAL
- PERIPHERAL NEUROPATHOLOGY
- Diagnosis
- Mimics
Statistics from Altmetric.com
Definition and epidemiology of CIDP
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is clinically defined as a ‘chronically progressive, stepwise or recurrent proximal and distal weakness and sensory dysfunction of all extremities, developing over at least 2 months, with absent or reduced tendon reflexes in all limbs and sometimes with cranial nerve involvement’.1 Sensory dysfunction is frequently present, most usually affecting joint position and vibration submodalities. Wasting is not prominent early in the disease. There are atypical forms, such as multifocal acquired demyelinating sensory and motor neuropathy (MADSAM, or Lewis–Sumner syndrome), pure sensory or pure motor CIDP and focal or distal forms (distal acquired demyelinating sensory polyneuropathy (DADS)).
A recent study from South-East England estimated the standardised prevalence rate of CIDP at 3 per 100 000, with a higher prevalence in males (range of estimated prevalence 0.8–8.9 per 100 000 in the previous eight studies).2 Most people with CIDP have a progressive rather than a spontaneously relapsing and remitting course, with a variable balance between motor and sensory symptoms.
The American Academy of Neurology established diagnostic research criteria for CIDP in 1991.3 There have since been further more practical diagnostic consensus guidelines from the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS).1 These and other proposed criteria typically comprise a combination of clinical and electrophysiological features (there have been 15 formal sets of published electrophysiological criteria for the diagnosis of CIDP4). Cases are classified as definite, probable or possible, depending upon the number of criteria fulfilled. In most, finding a raised CSF protein without CSF leucocytosis5 further supports the diagnosis. Clear evidence of macrophage-associated demyelination and remyelination, with or without a T-cell inflammatory endoneurial infiltrate in a sensory nerve biopsy, remains the gold standard supportive criterion (figure 1). There have been several validations of these diagnostic criteria, though none is 100% sensitive or specific1 ,3; for example, the EFNS/PNS criteria1 show a positive predictive value of 97% and negative predictive value of 92%,6 and different validation cohorts give widely varying sensitivities and specificities.5
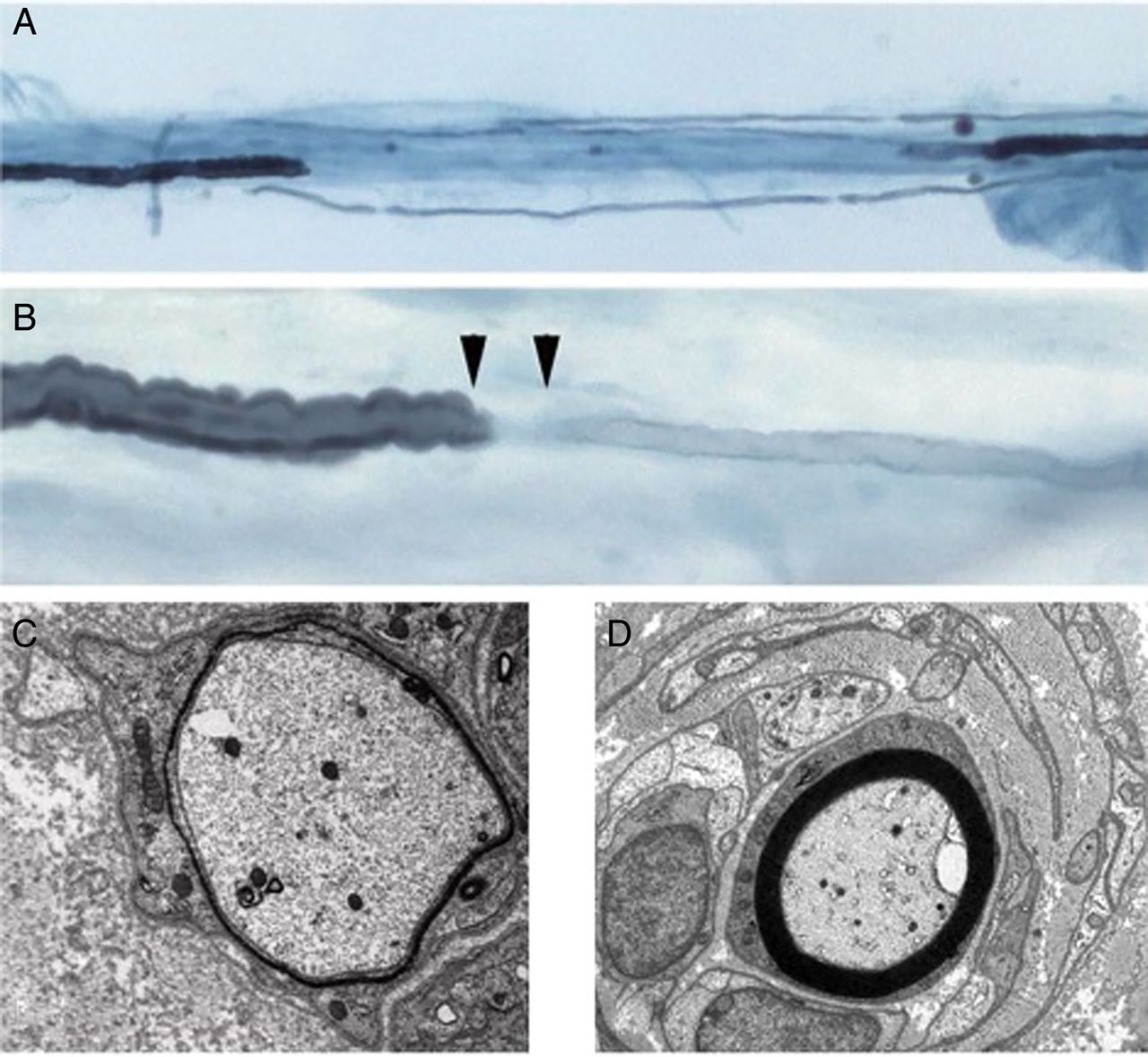
Demyelination in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): teased nerve fibres demonstrating a segment of demyelination (A); a widened node of Ranvier with a remyelinated segment (B); electron microscopy of a demyelinating fibre (C), and an onion bulb (D).
CIDP diagnosis in practice
Most CIDP cases are relatively straightforward to diagnose. In practice, a clinical diagnosis of CIDP is made in people with
-
a subacute presentation of patchy motor weakness and/or positive sensory symptoms (paraesthesia);
-
of at least 8 weeks duration;
-
with electrophysiological evidence of demyelination on nerve conduction studies;
-
with or without a raised CSF protein;
-
in whom alternative diagnoses, particularly hereditary neuropathies have been excluded.
Chameleons: atypical presentations of CIDP
While the diagnosis of CIDP is straightforward in typical presentations with excellent expert agreement, there is less universal diagnostic consensus in atypical presentations. This is reflected by the EFNS/PNS guidelines subdivision into definite, probable and possible CIDP cases, based on the clinical presentations.1 In a review of four typical and seven atypical cases of definite CIDP, a panel of 32 experts from 22 different centres confirmed CIDP in 97% of the typical cases, but in only 60% of the atypical cases.7
Atypical presentations with pure motor or sensory variants, focal, monomelic or asymmetrical variants in the context of comorbidities increase the likelihood of an initial alternative diagnosis.
In our practice, patients with CIDP occasionally have previously received a different diagnosis. With retrospect, it is usually possible to identify the point at which, and why, that erroneous diagnosis was made. Sometimes it is entirely justifiable (see Guillain–Barré syndrome below). Box 1 lists potential alternative diagnoses that turn out to be CIDP.
CIDP chameleons. Potential initial misdiagnoses in atypical presentations of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP)
-
Guillain–Barré syndrome
-
Motor neurone disease
-
Focal compression neuropathies (particularly ulnar and median compression)
-
Charcot–Marie–Tooth 1X (CMT1X)
-
Idiopathic sensory neuropathy
-
Sensory neuronopathy
-
Sensory polyradiculopathy
-
Diabetic neuropathy
-
Neuropathy secondary to amyloidosis
A diagnosis of CIDP cannot be made until symptoms have progressed for over 8 weeks. Some CIDP presentations are very acute (acute-CIDP or a-CIDP); a diagnosis of Guillain–Barré syndrome may entirely reasonably later be revised to CIDP. Incidentally, there may be a biological explanation for this, as such cases may show a persistent Fas deficiency in T-cells.8
If symptoms are confined focally to a single limb (monomelic involvement), or even to a single nerve, they may be initially thought to be a focal compression neuropathy, such as carpal tunnel syndrome or an ulnar neuropathy—and should be considered when patients have had unsuccessful decompression surgery. This CIDP chameleon is more likely when there is limited electrophysiology, directed solely at a carpal tunnel or ulnar nerve at the clinician's request, as it often is in technician-run ‘carpal tunnel diagnostic services’. Occasionally there may be no positive electrophysiological criteria for peripheral nerve demyelination at a more comprehensive first examination, but the clinician's index of suspicion or progression provokes a repeat examination.
Similarly, pure sensory variants of CIDP may be misdiagnosed as an idiopathic sensory axonal neuropathy, a sensory neuronopathy or a sensory polyradiculopathy. Symptoms may be attributed to a pre-existing condition, such as diabetes mellitus or amyloidosis. Idiopathic sensory axonal neuropathies progress linearly and slowly, and are seldom disabling. Diabetes mellitus does not usually cause a neuropathy unless severe or longstanding or both, and a significant neuropathy in a patient with mild diabetes should make one consider an alternative diagnosis. Amyloid is likely to have prominent pain and associated autonomic dysfunction, both features that would be against a diagnosis of CIDP.
Pure motor variants of CIDP may have a ‘motor neurone disease’-like presentation, particularly if the patient reports fasciculation and cramp, though will lack upper motor neurone signs and should be distinguishable with neurophysiology. Sensory abnormalities in the nerve conduction studies push a diagnosis of multifocal motor neuropathy towards MADSAM or CIDP, when motor conduction blocks and dispersion coexist.
Electrophysiological evidence of patchy temporal dispersion or conduction block (figure 2) in the context of a motor and/or sensory demyelinating polyneuropathy helps to distinguish CIDP from the hereditary demyelinating neuropathies, although dispersion and block can occur in some of the hereditary neuropathies, such as Charcot–Marie–Tooth-1X (CMT-X, see below), leading to diagnostic uncertainty. Nevertheless, clinicians often treat cases where there is diagnostic uncertainty as CIDP in the hope that there will be some clinical improvement. A diagnosis of CMT that turns out to be CIDP is unusual, as genetic diagnosis is so much easier in 2014.

Conduction block from the elbow to the axilla in the median nerve with slowing in all segments (reduction in amplitude and area) in a patient with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
CIDP mimics
The differential diagnosis of a chronic acquired demyelinating polyneuropathy is relatively short, but there are several CIDP mimics that should not be missed. Clinicians should be aware of the red flags that should prompt re-evaluation of the diagnosis of CIDP. Box 2 lists the red flags that should prompt one to consider an alternative diagnosis, while box 3 lists the features that positively support a diagnosis of CIDP.
Red flags
Red flags for the diagnosis of typical chronic inflammatory demyelinating polyradiculoneuropathy (CIDP):
-
Symptoms in the history
-
Background history of a slowly progressive neuropathy
-
‘Prominent’ pain symptoms, or significant early muscle aching
-
No (or minimal) symptomatic sensory disturbance
-
Comorbid conditions or other systemic features (oedema, weight loss, organomegaly, skin or nail pigmentation, rash, or gynaecomastia)
-
-
Signs in the examination
-
Cranial nerve involvement or papilloedema (figure 3)
-
Significant lower motor neurone wasting, especially in early disease
-
Respiratory muscle involvement
-
Head drop
-
Significant relative dominant hand weakness with median/ulnar separation*
-
-
Signs or investigation findings
-
Autonomic involvement
-
Sphincter disturbance
-
-
Anomalous response to treatment
-
Failure to respond to adequate and reasonable trials of treatment
-
Rapid and continued progression of symptoms
-
*Relative weakness of the dominant hand with median/ulnar separation suggests an alternative diagnosis, such as CMT1X[9] but is not of itself diagnostic of any single condition.
Features supporting a diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP)
Features supporting CIDP*
-
Elevated CSF protein with CSF white cell count <10/mm3
-
MRI evidence of hypertrophy or enhancement of the nerve roots (cervical, brachial, lumbrosacral or cauda equina)
-
Abnormal sensory electrophysiology in at least one nerve
-
Objective clinical improvement following immunomodulatory therapy
-
Unequivocal evidence of demyelination and/or remyelination by electron microscopy or teased fibre analysis in nerve biopsy† (figure 1)
*Modified from box 5 in the EFNS/ Peripheral Nerve Society (PNS) CIDP guidelines.1
†Other features on biopsy that support a diagnosis of CIDP include direct visualisation of inflammatory infiltrates of T cells and macrophage in association with demyelination.
Unusual symptoms and historical findings
Prominent pain is unusual in CIDP, and significant pain in the context of a mononeuritis multiplex, even with demyelinating features on electrophysiology, suggests a vascultic neuropathy. Significant calf muscle aching is often an early feature of POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes) and should prompt a search for other features of the syndrome, including mandatory serum paraprotein.
Other features in the history, which typically should exclude a diagnosis of CIDP at onset, include an associated fever or known intercurrent infections (Lyme disease, diphtheria) or drug or toxin exposure known to potentially cause neuropathy (hexacarbons, amiodarone). Clearly, a family history should be diligently sought (this often takes more effort than one would think) as this entirely changes the diagnostic pathway.
Unusual signs
The presence of upper motor neurone signs, prominent sphincter or respiratory involvement should prompt the clinician to consider an alternative or secondary diagnosis. Upper motor neurone signs do not occur in CIDP, but second diagnoses, such as cervical spondylosis, are not infrequent. There have been cases of coexisting myasthenia gravis.
Persistent asymmetrical hand weakness with a weaker dominant hand and greater median than ulnar nerve involvement in the dominant hand (weaker abductor pollicis brevis compared to first dorsal interosseous), in the context of poor treatment response, and especially if there is a long slowly progressive history, suggest an alternative diagnosis of Charcot–Marie–Tooth disease IX (CMT-X).9 There is often a clue in the correlated electrophysiology, which may also show the same pattern, as well as associated dispersion and block. Finding dispersion and block may lead to an initial inflammatory diagnosis.
Unusual investigation findings
Early significant axonal involvement is unusual, identified either on examination by early wasting or fasciculation or on nerve conduction studies. Even in the context of prominent demyelinating features, if there is a background of confirmed diabetes mellitus, then axonal loss is more likely to be from diabetic neuropathy rather than from CIDP. Early reports of an epidemiological causal association between CIDP and diabetes mellitus appear to have been incorrect.10
An IgM monoclonal gammopathy may be associated with high titre antibodies to myelin-associated glycoprotein (MAG).1 An anti-MAG antibody without the ‘typical’ phenotype of anti-MAG neuropathy may not be relevant although may be associated with unusual pathologies. We have seen amyloid deposition and vasculitis (sometimes with a mixed cryoglobulinaemia) caused by IgM paraproteins, without the ‘typical’ phenotype. Most relevant anti-MAG antibodies are reported as ‘strong positive’ on the current widely used Buhlmann ELISA.
Autonomic involvement
In contrast to Guillain–Barré syndrome, CIDP rarely has autonomic features. When present, autonomic dysfunction is never significant and mainly manifests as bowel and bladder complaints. Baroreflex-mediated peripheral vasoconstriction is relatively spared in CIDP and therefore significant orthostatic hypotension is uncommon.11 Any significant autonomic features should lead one to consider alternative diagnoses, especially amyloid, diabetic neuropathy, and rarely vasculitis or even a second diagnosis.
The efficacy of immune-modulatory therapies in CIDP
Four recent Cochrane reviews have evaluated the efficacy of treatments in CIDP,12–15 which include the three standard therapies. The evidence for the efficacy of corticosteroids is based on two randomised controlled trials, with patients treated with corticosteroids having improved neuropathy impairment scores after 12 weeks compared to patients randomised to no treatment. There was no statistical difference in remission rates at 1 year in people treated with standard dose oral prednisolone compared to monthly high-dose oral dexamethasone.12 Nevertheless, oral corticosteroids are typically employed as first-line treatment in cases of newly diagnosed CIDP. The quality of the evidence for the efficacy for intravenous immunoglobulin and plasma exchange is moderate to high, with plasma exchange providing significant short-term improvements in disability, clinical impairment and motor nerve conduction velocities, although there may be rapid deterioration postexchange.13 The evidence for intravenous immunoglobulin from the eight published randomised controlled trials confirms that intravenous immunoglobulin improves disability for at least 2–6 weeks compared to placebo, with comparable efficacy to plasma exchange and oral corticosteroids.14 Only one study (the ICE trial) had long-term follow-up and reported that the benefit of intravenous immunoglobulin on disability persisted for 24, and possibly 48 weeks, compared to placebo.16 There was no significant difference in the mean disability scores in people treated with intravenous immunoglobulin or plasma exchange at 6 weeks.17 Similarly, there was no difference in improvement in disability between people treated with prednisolone and intravenous immunoglobulin at 6 weeks.14 There was no significant difference in the frequency of side effects between the three treatment modalities although a more recent randomised controlled trial found that people treated with a 6-month course of intravenous immunoglobulin were less likely to stop treatment because of inefficacy, intolerance or adverse effects compared to treatment with intravenous methylprednisolone.18 Intravenous methylprednisolone, however, produced a more sustained remission when stopped, if it were tolerated for the full course.
Unusual response to immunotherapy
Seventy per cent of patients respond to one or another of the three standard therapies, and probably 90% respond overall.5 A failure to respond to treatment, or rapid deterioration of symptoms despite an adequate trial of treatment with at least two treatment modalities, should prompt a re-evaluation of the diagnosis.
Motor-predominant forms of CIDP present with clinical motor involvement only, but sensory involvement on electrical studies. Motor CIDP often behaves more like multifocal motor neuropathy with conduction block; these cases may worsen with corticosteroid treatment, and more rarely plasma exchange, but usually respond to treatment with intravenous immunoglobulin. Other conditions, such as MADSAM (Lewis–Sumner syndrome), chronic inflammatory sensory polyneuropathy, DADS and relapsing sensory ataxic neuropathy, are all considered atypical variants of classical CIDP. Although the clinician may be fortunate in treating such patients effectively, atypical variants have a somewhat unpredictable and typically poorer treatment response compared to typical CIDP.5 A careful clinical history and examination and use of appropriately comprehensive electrical studies can differentiate these clinically important variants. The DADS acronym is confusingly also applied to anti-MAG neuropathies (see below), and this may be because some laboratory tests in current use are unable to detect all anti-MAG antibodies.
Occam's razor, Hickam's dictum and Crabtree's bludgeon
When considering any neurological diagnosis, not least CIDP, it is practical and useful to remain in touch with Occam's razor. Occam's razor can be summarised as ‘entities must not be multiplied beyond necessity’,19 which in medical terms can be paraphrased as ‘when investigating a patient with multiple symptoms, a single unifying diagnosis should be sought rather than two or more unrelated ones’. A singular unifying diagnosis is not always possible, but the application of Hickam's dictum (multiple symptoms and signs may be due to more than one condition, or more commonly ‘a man can have as many diseases as he damn well pleases’20) should be infrequent under the age of 80 years. One should always be aware, and be wary, of applying Crabtree's bludgeon (‘no set of mutually inconsistent observations can exist for which some human intellect cannot conceive a coherent explanation, however complicated’21), if applied, should prompt the clinician to step back and re-evaluate Occam. Usually, the diagnosis is straightforward and unifying, and the disease will behave like it is supposed to.22
Potential alternative diagnoses
So what are the alternatives? Box 4 lists some genetic and acquired conditions that can lead to diagnostic confusion.
Potential genetic and acquired chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) mimics
Alternative diagnoses to consider in treatment failure CIDP
-
Genetic Mimics
-
GJB1 mutations (CMT1X)
-
Transthyretin familial amyloid polyneuropathy (TTR-FAP)
-
CMT 4C (SH3TC2 mutations)
-
CMT4J (FIG 4)
-
HSAN1 (SPTLC1)
-
CMT1A (homogenous slowing)
-
CMT1b
-
HNPP
-
GDAP1
-
MNGIE and rare mitochondrial disorders
-
-
Acquired Mimics
-
POEMS
-
Paraprotein related (MGUS/WM/lymphoma) (IgM associated)
-
AL amyloid
-
Treated-related fluctuations in GBS (GBS-TRF)
-
Diabetic neuropathy/Diabetic lumbro-sacral radiculoplexus neuropathy (DLRPN)
-
Vasculitis
-
POEMS: polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes
Genetic mimics
Possibly the most common genetic CIDP misdiagnosis is CMT-X.23 This is caused by one of a large number of mutations in the GJB1 gene, which encodes for the gap junction protein connexion-32. CMT-X is often electrically patchy and can sometimes have evidence of temporal dispersion and conduction block on nerve conduction studies (unlike most other forms of CMT) (figure 2). The lack of response to treatment and minimal electrophysiological progression over time should lead one to suspect the diagnosis. One might elicit a X-linked family history with detailed questioning, and sometimes with the examination of mildly affected and asymptomatic female relatives. Recessive demyelinating neuropathies are uncommon but need to be kept in mind even if there is an equivocal response to intravenous immunoglobulin. Even when there is a recognised genetic diagnosis, the condition may be ascribed to CMT1A with a superimposed inflammatory polyneuropathy (Hickam's dictum). Cases with CMT4C (SH3TC2, previously KIAA1985)24 usually have early onset in the first or second decade, delayed or difficulty walking, and often cranial nerve involvement (figure 3) and highly specific features on electron microscopy of longitudinally extended Schwann cell cytoplasmic processes on peripheral nerve biopsy.24
CMT4J (figure 4)25 presents with rapid progressive asymmetrical weakness, and a demyelinating neuropathy. Both have been misdiagnosed with CMT1A and CIDP on occasion and, therefore, should be considered in the differential diagnoses in cases of treatment failure.

Thickened 3rd nerves in a patient with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
Recently, there has been a description of a new recessive phenotype (autosomal recessive spastic ataxia of Chavenoix–Sauvigny or ARSACS), associating a demyelinating neuropathy with ataxia and spasticity: most of these patients have a mutation in sacsin. Although spasticity and cerebellar features are usually prominent, there are phenotypic variants with only neuropathy.26 A nerve biopsy may contain significant numbers of lipofuscin granules.
Another important genetic diagnosis is transthyretin familial amyloid polyneuropathy (TTR-FAP),27 particularly in cases of sporadic (non-familial) TTR-FAP.28 TTR-familial amyloid polyneuropathy is an autosomal dominant genetic condition with multisystem involvement. It is caused by one of several mutations in the transthyretin gene with the V30M mutation being the most common mutation in people with Portuguese ancestry.28 The classical presentation is one with a length-dependent sensorimotor predominantly small-fibre polyneuropathy (although some may have some demyelinating features), and autonomic dysfunction (postural hypotension intermittent diarrhoea, weight loss and impotence). There are other rare non-TTR forms of familial amyloid polyneuropathy. The diagnosis is typically confirmed on biopsy of nerve, rectum or abdominal fat.27 ,28 There is an easily available genetic test for TTR mutations, which can be performed without resorting to biopsy if the diagnosis is suspected.
MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) can occasionally present with a demyelinating peripheral neuropathy and be misdiagnosed as CIDP.29 In such cases, neuroimaging (looking for leucoencephalopathy), testing urinary thymidine and uridine, and screening for thymidine phosphorylase (TYMP) mutations, should be considered, particularly if there is subsequent development of prominent ocular (ptosis, external ophthalmoplegia) or gastrointestinal symptoms.30
Acquired mimics
Among the potential acquired diagnoses to consider is that of the POEMS syndrome. While the diagnosis of POEMS syndrome in its advanced presentation is not difficult, up to 50% present with an initial, even acute, (demyelinating or mixed demyelinating/axonal) polyneuropathy,31 ,32 leading to a diagnosis of CIDP.
Features that help distinguish the polyneuropathy of POEMS from that of CIDP include a distal dominant painful polyneuropathy (prominent neuropathic foot pain, or even less severe but typical calf aching), more frequent muscle atrophy, distal dominant lower limb weakness31 and more rapid onset of inability to walk independently (median period of 9.5 months from onset).32
A paraprotein (or monoclonal lymphoproliferative disorder) is an essential major criterion in the diagnosis (Dispenzieri criteria), and may only be discovered with a serum immunofixation, Bence Jones protein, or serum-free light-chain analysis. Electrophysiological features that favour POEMS include the absence of conduction block (rare), predominant nerve conduction slowing in the intermediate rather than the distal nerve segments, and more severe involvement in the lower rather than the upper limbs.33 Clinical suspicion of POEMS should lead to measurement of serum level of vascular endothelial growth factor (VEGF), which is typically elevated and supports the diagnosis. Indeed, the presence of an elevated serum VEGF-A level of twice the upper limit of normal in the context of a demyelinating polyneuropathy and a paraprotein, and the absence of hypoxia or iron deficiency anaemia, is almost pathognomonic for POEMS syndrome and should prompt other appropriate investigations to support the diagnosis and to stage the disease.
Polyneuropathy with IgM monoclonal gammopathy of undetermined significance (IgM MGUSP) is an immune-mediated polyneuropathy in most cases. It is typically characterised by a predominantly distal and sensory impairment with about half having antimyelin-associated glycoprotein (anti-MAG) antibodies.34 Electrophysiologically prolonged distal motor latencies often occur in IgM MGUSP, which helps distinguish it from CIDP. Making this diagnosis is important, as all people with IgM MGUSP should be investigated for a malignant blood dyscrasia.34 In people with an IgM MGUSP, a later age of onset and demyelinating features are associated with a worse prognosis, while the presence of anti-MAG antibodies suggests a better prognosis.35
Primary AL amyloidosis is an acquired condition characterised by abnormal proliferation and deposition of monoclonal immunoglobulin light chain in tissues; 15% have a dominant peripheral neuropathy.36 When there is a peripheral neuropathy in the context of primary AL amyloidosis, this is typically predominantly a symmetrical sensory neuropathy with minimal motor involvement. Moreover, autonomic features are often prominent in addition to systemic symptoms, such as unexplained weight loss, fatigue and constipation. Features that may not be prominent at presentation, but develop subsequently and suggest primary AL amyloidosis, include non-diabetic nephrotic syndrome, non-ischaemic cardiomyopathy, hepatomegaly or elevated alkaline phosphatase level with normal imaging.37
Almost all patients with primary amyloidosis have an elevated level of the responsible free light chain, underlying the importance of detailed immunofixation of serum or urine.
Another alternative diagnosis is treatment-related fluctuation in GBS (GBS-TRF), reported in 8–16% of cases of GBS.38 The treatment strategy and long-term prognosis differ considerably between the two conditions. In differentiating GBS-TRF from acute-onset CIDP, a diagnosis of acute CIDP should be considered in a person previously diagnosed with GBS if there is clinical deterioration more than 9 weeks after onset, or if clinical deterioration occurs on three or more occasions.39
Diabetic neuropathy may occasionally have a demyelinating pattern and meet the electrophysiological criteria for CIDP, resulting in diagnostic confusion.40 Similarly vasculitic neuropathy may rarely be mistaken for CIDP with the diagnosis only confirmed after a nerve biopsy.
Second-line investigations
Box 5 gives a summary of the second-line investigations to detect mimics, or for cases of treatment failure. The clinician should first return to the history with a view to discovering any suggestion of a family or developmental history. A detailed family history should be obtained with particular focus on ethnicity (Irish/Portuguese/Mediterranean), which may suggest FAP. Other aspects in the history, such as the revelation of persistent ‘toe walking’, poor athletic performance during school years, difficulty in shoe fitting as a child, or failure to be able to wear high heels, are quite revealing. These features may require active history taking, and suggest a more chronic course and favour a genetic rather than an acquired aetiology. Nerve conduction studies should be repeated looking for evidence of rapid electrophysiological progression; alternatively, these may show complete stability, suggesting a genetic cause. Serum protein electrophoresis has only a 60% sensitivity for detecting paraproteins in the context of neuropathy (immunoparesis is rare) so immunofixation (95% sensitivity) and urinary Bence Jones protein should be performed, and serum-free light-chain analysis (a few additional cases) considered. MR scanning of the brachial and lumbrosacral plexuses with gadolinium should be requested looking for evidence of nerve root or plexus hypertrophy or enhancement (figure 4).41 Useful sequences are STIR and T1 weighted images, with and without gadolinium, but data continue to emerge on the specificity and sensitivity of MRI findings.42 In cases where there has been no significant deterioration over time either clinically or electrophysiologically, it is reasonable to consider sequencing of the GJB1 gene, or an alternative genetic diagnosis.
Second-line investigations to be considered in treatment failure chronic inflammatory demyelinating polyradiculoneuropathy (CIDP)
Second-line investigations in treatment failure CIDP
-
Detailed family history (including ethnicity)
-
Developmental history
-
Repeat nerve conduction studies
-
Repeat serum electrophoresis with immunofixation, serum-free light-chains and Bence Jones protein
-
Serum vascular endothelial growth factor (VEGF)
-
Nerve root/plexus/peripheral nerve imaging (MRI/U/S)
-
GJB1 gene sequencing or other genetic requests
-
Nerve biopsy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Spinal root hypertrophy in a patient with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): Sagittal T2-weighted MRI of the cervical (A) and lumbosacral (B) spine with axial images at the level of L4 (C) and S1 (D) demonstrating extensive and diffuse hypertrophy of the spinal nerve roots. There is associated enlargement of the spinal canal and posterior scalloping of the vertebral bodies. Hypertrophy of the lumbosacral plexus bilaterally is also shown (arrows). Note the multilevel vertebral insufficiency fractures from chronic prednisolone treatment.41
A nerve biopsy generally remains a last resort in diagnosis, but should definitely be considered, but not always performed, in all cases of treatment failure, particularly if there are other red flags (box 1). Ideally, the biopsy should be performed in a centre with surgeons and neuropathologists experienced in nerve biopsies and processing. The target should be a sensory nerve with evidence of involvement on nerve conduction studies (sural nerve, superficial peroneal or, more rarely, the superficial radial or the dorsal ulnar nerves). Typical findings of CIDP on biopsy include an inflammatory infiltrate of T cells (>5 lymphocytes per fascicule) and macrophages in the endoneurium; unequivocal evidence of demyelination and remyelination is far less common5, although much more convincing for CIDP. Teased fibre analysis and cross-sectional and longitudinal electron microscopy are very valuable.1 (figure 1). Particular attention should be paid for evidence of vasculitis or amyloid deposition, with the biopsy sent to a dedicated neuromuscular pathologist if appropriate. There are several techniques of Congo red staining, some more successful than others.43 Additionally, a full panel of immunohistochemical inflammatory markers is essential.
In cases where atypical POEMS syndrome is suspected, serum VEGF should be measured, a sural nerve biopsy considered in addition to body imaging, looking for evidence of organomegaly, and blood tests for evidence of endocrine involvement. Investigations for an underlying malignancy, including CT/positron emission tomography (PET), should be initiated in cases of rapid progression or non-response to treatment.
Recent research using MRI to evaluate sciatic nerve cross-sectional area can help to differentiate between inherited and inflammatory neuropathies.44 While still a research tool at present, it offers the potential of an objective pathological marker to complement clinical investigations in cases of diagnostic uncertainty.
Other treatment options/regimens
Ultimately, most cases of treatment failure retain a diagnosis of CIDP, as 70% of people with CIDP respond to any one of the first-line treatments with corticosteroids, intravenous immunoglobulins, or plasma exchange, with a further 10% responding to a combination of treatments.1 ,5 Interestingly, while 80% respond to one of the first-line treatments,5 people with a monophasic or a relapsing–remitting course (as opposed to a chronic progressive course) or a greater than twofold CSF protein increase (≥0.84 g/L) show a better response to treatment with intravenous immunoglobulin.6 However, overall, 10% of people with definite CIDP will be in the treatment failure group.
While the management of refractory CIDP is not the primary focus of this article and is covered in much greater depth elsewhere,45 brief mention of therapeutic strategies employed at our institution is appropriate. In cases where people with a secure diagnosis of CIDP fail to improve following a combination of treatment with corticosteroids, intravenous immunoglobulin and plasma exchange, combination therapy with plasma exchange followed by intravenous immunoglobulin may be trialled. Oral immunosuppression (methotrexate, azathioprine, mycophenolate mofetil, or ciclosporin) is often used despite lack of controlled trial evidence. Failing this, consideration is given to the use of pulsed cyclophosphamide (Cyclops regimen)46 or rituximab,15 although high-quality data are lacking, other than in case reports and series. Other treatment options, reported as single case reports or small case series, include interferons (α and β), alemtuzemab, etanercept, fingolimod and stem cell transplantation.15
Conclusions/summary
We have tried to highlight the clinical and electrophysiological features that should make one consider an alternative diagnosis in cases where CIDP fails to respond to appropriate standard therapy. In particular, we have considered potential alternative diagnoses and features that help distinguish such conditions from CIDP.
We have also considered the CIDP chameleons that may be initially misdiagnosed in atypical presentations. In the end, in most cases of treatment failure, a diagnosis of CIDP will be retained, but it is important to consider alternative diagnoses, particularly when different treatment regimens may be required.
Acknowledgments
We wish to thank Professor Sebastian Brandner for kindly providing us with the images of the nerve biopsy. We also wish to thank Dr Zane Jaunmuktane for her helpful comments on the different methods for staining of amyloid on nerve biopsy.
References
Footnotes
-
Contributors All authors were involved in the concept of the study, drafting of and approval of the final manuscript.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Haider Katifi, Southampton, UK.
Linked Articles
- Editors' choice
Other content recommended for you
- Comparison of the diagnostic accuracy of the 2021 EAN/PNS and 2010 EFNS/PNS diagnostic criteria for chronic inflammatory demyelinating polyradiculoneuropathy
- Chronic inflammatory demyelinating polyradiculoneuropathy in solid organ transplant recipients: a prospective study
- Unclassified clinical presentations of chronic inflammatory demyelinating polyradiculoneuropathy
- Atypical CIDP: diagnostic criteria, progression and treatment response. Data from the Italian CIDP Database
- CSF sphingomyelin: a new biomarker of demyelination in the diagnosis and management of CIDP and GBS
- Frequency of diabetes and other comorbidities in chronic inflammatory demyelinating polyradiculoneuropathy and their impact on clinical presentation and response to therapy
- Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype
- Recommendations on diagnostic strategies for chronic inflammatory demyelinating polyradiculoneuropathy
- Oligoclonal IgG bands in chronic inflammatory polyradiculoneuropathies
- MRI of the cervical nerve roots in the diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy: a single-institution, retrospective case–control study