Article Text

Abstract
The diagnosis of acute intermittent porphyria (AIP) is often overlooked. We describe a patient with this condition who had all the ‘bells and whistles’, in whom the diagnosis was only made after considerable delay. Far from an esoteric condition haunting examination candidates, AIP is an important cause of a broad spectrum of neurological symptoms. Its early recognition allows the astute clinician to prevent potentially devastating sequelae. We provide practical guidance on the investigation and management of this complex disorder. With a ‘back to basics’ approach to the underlying genetics and biochemistry, we hope to dispel some of the confusion that may obstruct a timely diagnosis.
- porphyria
- neuropathy
Statistics from Altmetric.com
Introduction
A 42-year-old woman presented to a district general hospital with abdominal pain and vomiting. For 3 years she had repeatedly attended her general practitioner with daily vomiting, but with no cause found. Two weeks before this presentation, she had returned from a week’s holiday in Spain; since her return, the vomiting had become much more frequent (up to 7 times per day). Before the holiday, she had started a progesterone-only oral contraceptive for heavy periods. On assessment in the emergency department, she had mild hypertension but appeared otherwise well. Following intravenous fluids and ondansetron, she was discharged.
Two days later, she re-presented after three seizures. One was witnessed by ambulance staff who described a generalised tonic–clonic seizure that terminated following lorazepam. Her seizures were attributed to alcohol withdrawal, although with no basis for this assumption, and she refuted it vehemently. A CT scan of head was normal, but her peripheral white cell count was elevated at 16×109/L (3.6–11.0), predominantly a neutrophilia at 12.5×109/ L (1.8–7.5). She was started on Pabrinex and phenytoin but during her 5-day admission she had four further seizures. She was referred to the first seizure clinic and discharged.
Five days later she was readmitted, this time with severe abdominal pain and feeling generally unwell. She was discharged the same day with a diagnosis of constipation, attributed to using codeine for abdominal pain.
Six days later she was admitted for the fourth time, with profound lethargy and inability to cope at home. On examination, she had a marked tachycardia at 130 (sinus rhythm) and mild abdominal tenderness but with no other signs. Blood tests were normal apart from a new hyponatraemia of 129 mmol/L (135–145) and a mildly elevated white cell count of 14.4×109/L. Two days later she developed proximal muscle pain and weakness; examination showed moderate bilateral hip flexion weakness. Her serum creatine kinase concentration was normal. Over the next few days she developed profound weakness in all four limbs.
Twenty-five days after her initial neurological presentation, she was transferred to our department with a probable diagnosis of Guillain–Barré syndrome. On admission, she had marked bilateral weakness, which was predominantly proximal. She had no movement at the shoulders or elbows and mild weakness at, and distal to, the wrists. The lower limbs were similarly affected, with no movement in all movements proximal to the ankle and mild weakness distal to the ankle. Sensation was normal. The deep tendon reflexes were normal, with flexor plantar responses. Cranial nerve examination, including eye movements, was normal. The purely motor involvement and proximal distribution of weakness were not in keeping with Guillain–Barré syndrome.
Cerebrospinal fluid (CSF) examination was normal, with no cytoalbuminologic dissociation. She was persistently hypertensive.
Neurophysiology showed a severe axonal motor neuropathy with proximal axonal damage. The neurophysiologist was struck by the patient’s tearful, almost hysterical demeanour.
The combination of abdominal symptoms, seizures, axonal neuropathy and behavioural abnormalities raised the suspicion of porphyria. Her urinary porphobilinogen/creatinine ratio was significantly elevated at 91.3 µmol/mmol (normal 0–1.5). Genetic analysis identified a pathogenic heterozygous mutation in the hydroxymethylbilane synthase (HMBS) gene (c.730_731delCT), confirming the diagnosis of acute intermittent porphyria (AIP). We started intravenous dextrose (4% dextrose, 1 L over 12 hours) and haem–arginate infusion (3 mg/kg diluted in 100 mL of normal saline, given over 30 min via a large cannula, followed by a saline flush). We replaced phenytoin with levetiracetam, as phenytoin induces hepatic enzymes that may precipitate porphyric attacks.
She remained an inpatient for 20 days, during which time her dysphagia and pain improved considerably. Her urinary porphobilinogen/creatinine ratio dropped to 35 µmol/mmol within 4 days of treatment. This reflects haem–arginate’s suppression of endogenous haem synthesis, but does not inform the prognosis. Her dysphagia and pain quickly resolved. She repeatedly reported improvements in her mood as well as her strength, though the observed improvements in power were modest initially, improving from complete flaccid paralysis to severe weakness in the arms and legs during admission. Twenty-eight months later, she could walk short distances, using a wheelchair for longer distances. On examination 3 years later, most muscle groups have returned to full strength but she remains severely impaired by remaining distal weakness and contractures. She no longer has pain and has not had further seizures. She still needs help for many activities of daily living but has had no further attacks. While this represents a marked improvement from presentation, porphyria-related neuropathy can be almost completely reversible if treated promptly, with most patients returning to walking within a year.
The delays in diagnosing her AIP may baffle some readers: surely, this was a case of porphyria with all the bells and whistles? Yes, but too many key aspects were initially not known, and others ignored or misinterpreted. The prolonged history of vomiting became known only after detailed review and the initial interpretation of alcohol withdrawal as the key trigger for her seizures was mere speculation. Her occasionally erratic behaviour on the ward combined with difficulty in obtaining a coherent history were misinterpreted as evidence of functional overlay but should also have helped with the diagnosis of porphyria.
The porphyrias are an uncommon, heterogeneous group of disorders that we may think of as more likely to be encountered in exams rather than on our wards. Those of relevance to the practising neurologist include AIP, variegate porphyria, hereditary coproporphyria and the exceptionally rare delta-aminolevulinic acid dehydrogenase deficiency porphyria. They can manifest unpredictably and are easily missed without a high degree of clinical vigilance. They occupy a challenging area of overlap in the Venn diagram between environmental, genetic and pharmacological factors.
Biochemistry
To understand the porphyrias, we must first discuss the haem synthesis pathway. Haem is synthesised in every nucleated cell of the body and comprises an iron molecule resting in a heterocyclic ring. About 85% of body’s haem is produced in the erythroid cells, incorporated into haemoglobin. Most of the remainder is synthesised in the liver and used in the production of the P450 enzymes. The haem synthesis pathway has eight enzymatically facilitated steps. Each is associated with an intermediate product, a porphyrinogen (or porphyrin precursor), and deficiency of each of these is associated with a clinical syndrome: the porphyrias. The first step of synthesis occurs within the mitochondria; the next four stages are carried out in the cytoplasm, before returning to the mitochondria for the final three steps (figure 1).1–3 The porphyrias can be divided into distinct groups, depending on different clinical, biochemical or genetic criteria (table 1).1–3
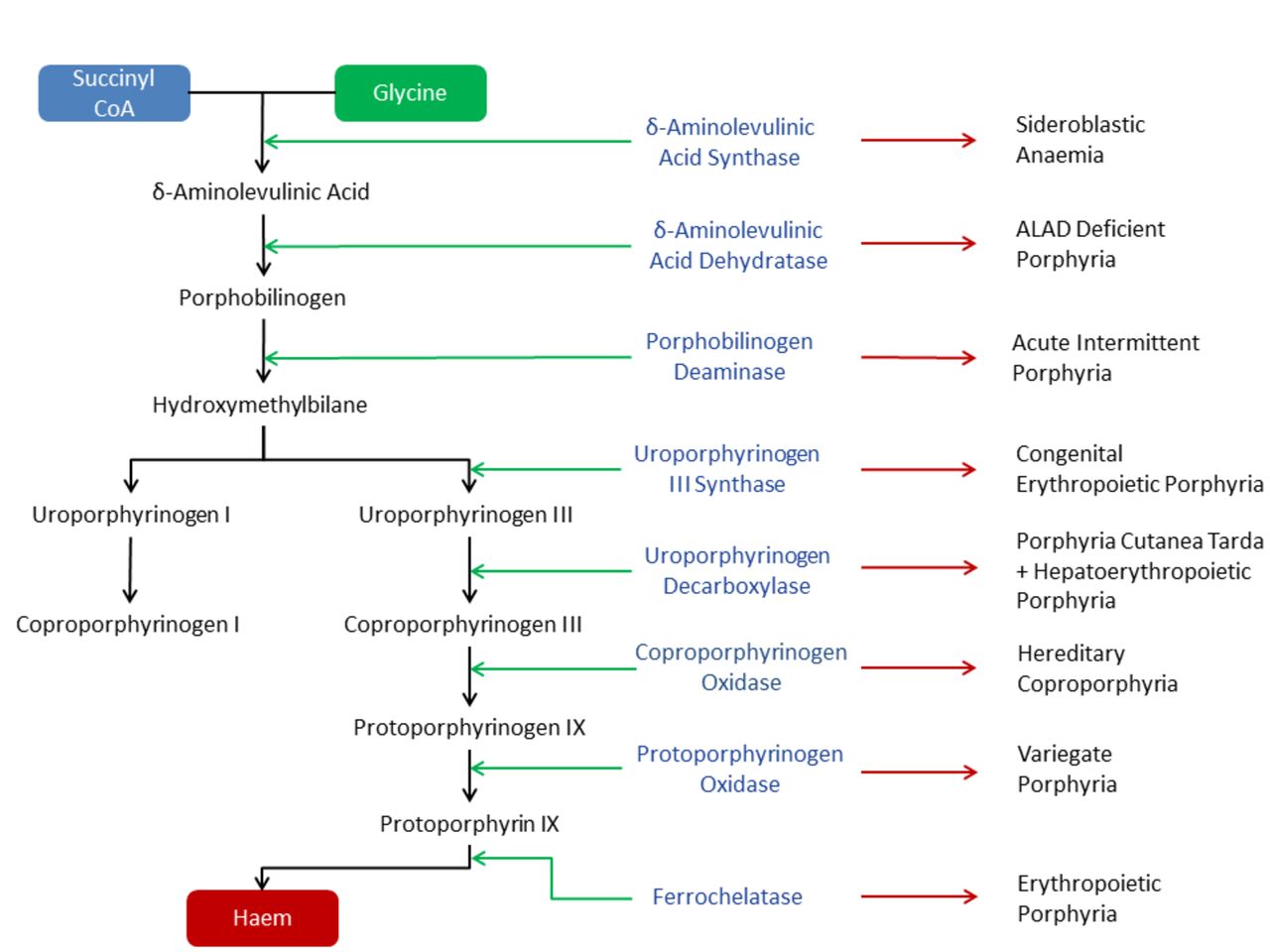
{kind=link}
Haem biosynthesis pathway. Green arrow—metabolic step facilitated by respective enzyme; red arrow— corresponding porphyria subtype. ALAD, Delta-aminolevulinic acid dehydratase.
Categories of porphyria
The porphyrias relevant to the neurologist
We propose a practical approach to the four acute porphyrias relevant to neurological practice. All acute porphyrias are characterised by neurovisceral phenomena that are absent in the exclusively cutaneous porphyrias. These lead to the familiar triad of abdominal pain, neuropsychiatric symptoms and neuropathy.1 3 They probably result from toxic effects of high delta-aminolevulinic acid concentrations on autonomic, peripheral and central nervous systems. Acute attacks are triggered by increased hepatic production of delta-aminolevulinic acid to produce more haem. Factors causing this increase include: medications, alcohol, endogenous steroids (explaining the relationship to puberty and the menstrual cycle) and starvation. The Drug Database for Acute Porphyrias provides an extensive list of drugs that must be used with caution in people with acute hepatic porphyrias.4 For neurologists, the most important aspect of pharmacotherapy in patients with porphyria is to avoid carbamazepine or phenytoin when treating seizures and to select gabapentin or levetiracetam instead. The mechanisms through which delta-aminolevulinic acid exerts its pathophysiological effect are complex. They are likely to include the propagation of free radicals within the central nervous system, the competition for gamma-aminobuytric acid (GABA)-binding sites, the impairment of mitochondrial function with a secondary inability to maintain axonal integrity and the interference of normal Na/K ATPase function, rendering the axonal membrane electrically unstable.3 5–8
Clinical features
AIP has varied clinical features. A study of 108 North American patients recorded the symptoms encountered during such an attack, which included:
Abdominal pain, nausea, vomiting and constipation
Anxiety and depression
Weakness
Hypertension and palpitations
Variegate porphyria and hereditary coproporphyria may present with acute attacks that are identical to those in AIP, although they may also present with light-sensitive skin lesions with or without attacks. Delta-aminolevulinic acid dehydratase deficiency is extremely rare, usually presenting in childhood with a chronic neuropathy or, in 40% of cases, acute attacks.9 We will now cover some particularly important neurological features of porphyria in greater detail.
The porphyric neuropathy
A peripheral neuropathy complicates 10–40% of acute episodes and most commonly occurs in AIP. It typically presents 3–75 days into an attack and reaches maximal intensity at an average of 2–4 weeks.3 7 The neuropathy is typically of acute axonal type. At its maximal severity, it can be so severe as to cause complete quadriplegia and respiratory failure, necessitating ventilatory support.7 It can be difficult to distinguish from other causes of an acute neuropathy such as the axonal variant of Guillain–Barré syndrome. However, some characteristic features can help in the differential diagnosis. The proximal muscles are predominantly affected (80%) and in half of the cases the weakness begins in the upper limbs. The distribution may also be asymmetric.7 10 There may be altered sensation in 60% of cases, typically a ‘bathing-trunk’ paraesthesia, with proximally altered sensation.3 The deep tendon reflexes, particularly the ankle jerks, may be retained, though some patients have global hyporeflexia. Profound myalgic pain may develop.11 Cranial nerve involvement accompanies 75% of cases of porphyric neuropathy and tends to develop after the limb and trunk symptoms. It may affect the facial and vagus nerves and less commonly the trigeminal, hypoglossal, accessory and oculomotor nerves.3
Porphyric neuropathy differs from Guillain–Barré syndrome in several ways:
There is a predilection for involving the proximal muscles and the upper limbs.
CSF during a porphyric attack usually does not show the classical cytoalbuminologic dissociation.
Electromyography typically shows an axonal motor neuropathy (rather than demyelinating neuropathy) with most marked denervation proximally.3 10 11
Seizures
Seizures occur in about 5% of patients with AIP. They are usually complex partial seizures, with or without secondary generalisation. However, some patients develop absences, myoclonic jerks and tonic–clonic seizures as well as epilepsia partialis continua.12 13
Hyponatraemia accompanying acute attacks may reduce the seizure threshold. Cortical laminar necrosis resulting from overzealous correction of serum sodium may act as a focal epileptogenic focus, propagating seizures even after an AIP attack.14
Encephalopathy develops in up to 70% of porphyric attacks.3 The clinical and radiological features of the porphyric encephalopathy may resemble posterior reversible encephalopathy syndrome, including high MRI-T2 signal areas in the occipital or frontoparietal cortices that do not enhance with contrast.15 Other abnormalities include the focal lesions (see above) and white matter lesions that can resemble the plaques of multiple sclerosis.16
Neuropsychiatric features
Twenty to fifty-eight per cent of patients with acute porphyria develop neuropsychiatric symptoms during the acute attacks.17 These may begin with minor behaviour changes, such as anxiety, impatience or insomnia.18 Other features include severe depression, anhedonia, grandiose delusions and severe psychotic episodes, resembling symptoms more typically associated with schizophrenia. They include both positive and negative features and can occur without neurological symptoms.11 19 Clinicians must be vigilant as some patients attempt suicide.20
Autonomic features
Over 90% of acute attacks are associated with typically severe and ill-localised abdominal pain.3 It may be so severe so as to necessitate exploratory surgery, with the risk of anaesthetics further exacerbating the porphyric attack.7 Autonomic features such as nausea, vomiting and constipation are also very common and typically dominate the early stage of an acute attack. Tachycardia and hypertension are also common, and there may be episodic sweating. Autonomic symptoms typically improve as the attack resolves.3
Diagnosis
Ultimately, the diagnosis of an acute porphyria attack relies on specific biochemical features. When suspecting an acute porphyric attack, the first step is analysis of urinary porphobilinogen in a random, light-protected sample. During an acute attack, there is a substantial increase in urinary porphobilinogen; clinicians should not overinterpret slight increases. Patients with an increased urinary porphobilinogen require further investigations. First, they need a repeat porphobilinogen together with porphyrins and delta-aminolevulinic acid, measured in the same urine sample, preferably at a reference centre. Second, they need porphyrin fluorescence emission scanning performed on a light-protected plasma sample. A plasma emission peak ≥624 nm is consistent with variegate porphyria. If there is no such peak, then the next step is faecal porphyrin analysis to distinguish between hereditary coproporphyria and AIP. If the ratio of coproporphyrin III to coproporphyrin I is above 1.5, the diagnosis is hereditary coproporphyria; if below 1.5, the diagnosis is AIP.21 This approach applies only to patients tested at the time of an acute attack. In remission, the urinary, faecal and plasma porphyrins are generally normal in the three autosomal dominant porphyrias. The plasma spectroscopy findings in variegate porphyria remain unchanged in remission.1 22–24 The challenges posed in the biochemical analysis of a patient in remission highlight the necessity for genetic testing in those at risk (see below). Usually an acute attack leads to identification of the proband case, with asymptomatic carriers of the genetic mutation in their families identified through screening.25 Low clinical penetrance is a recognised feature of the autosomal dominant acute porphyrias.2
Treatment
The principles of early management are to address any porphyrinogenic factors and to downregulate the haem synthetic pathway, thus reducing the amount of toxic delta-aminolevulinic acid produced.3 26
The cornerstone of specific treatment for an acute porphyric attack is intravenous haemin. The aim of treatment is to replace the body haem stores, thus downregulating hepatic production of delta-aminolevulinic acid. In Europe, haem–arginate (Normosang) is available. In the USA, lyophilised haematin is used. Early treatment with haem–arginate is associated with significantly improved outcome. It is normally given in doses of 3 mg/kg diluted in 100 mL of either saline or 20% albumin, into a large peripheral vein or central line, daily for 4 days. Saline flushes should be used to reduce the risk of phlebitis.27 Diluting the haem–arginate in 20% albumin rather than saline may reduce these irritant effects.28
Haem–arginate prevents further deterioration in porphyric neuropathy but does not reverse established nerve damage. Patients may relapse after the initial course and may require further courses until they are clinically stable.
In mild attacks, with no vomiting, weakness or hyponatraemia, it is worth trying a high carbohydrate diet with supportive measures for up to 48 hours before giving more specific treatment. If there are any neurological complications, however, patients need immediate and specific treatment (see table 2).28 Porphyrinogenic factors include medications, and in the case described above, progesterone may have contributed. Neurologists may find it particularly challenging to avoid these drugs and yet to control seizures, since many antiepileptic medications, especially phenytoin and carbamazepine, induce CYP450. Gabapentin and levetiracetam are the antiepileptic drugs of choice in this situation.3 27 29 30 Hyponatraemia complicates 40–90% of acute porphyric attacks,1 3 resulting from both the syndrome of inappropriate antidiuretic hormone secretion and a combination of renal and gastrointestinal salt loss.31 Hyponatraemia leads to symptoms at a serum concentration below 125 mmol/L, and seizures may develop at 112 mmol/L.14 When there is hyponatraemia, clinicians must take care when giving fluids; we recommend liaising closely with the endocrine team and monitoring in at least a Level 2 environment. Chronic hyponatraemia should be corrected only slowly (less than 6 mmol over 24 hours) to minimise the risk of central pontine myelinolysis.28
Clinical manifestations of an acute porphyric attack and respective symptomatic treatment
Table 2 summarises symptomatic treatment options.
Once the acute attack has remitted, the key aim is to prevent further episodes and to identify any other at-risk family members, who are typically asymptomatic, through genetic testing.25 Prevention is largely through education on avoiding potential precipitating factors. Patients with recurrent attacks may need prophylactic haem therapy to reduce the severity and frequency of their attacks.9 27 28 32 Some patients develop dependence and need their treatment increased from monthly to twice weekly. Long-term complications of recurrent haem therapy include loss of the superficial veins and iron overload (with its accompanying features).27 The first liver transplant for this indication was performed in 2002, in a 19-year-old woman with severe AIP. Since then, there have been further liver transplants for AIP, all resulting in complete resolution of the attacks.3 32–36
A note on porphyria and women
Hormonal fluctuations around menstruation can trigger recurrent attacks. The use of the oral contraceptive is contentious and requires individual assessment. Some women with acute porphyria can tolerate oral contraceptive pills without triggering acute attacks. However, oral contraceptives provoke acute attacks in 5–14% of women with AIP, which readily explains why guidelines for female patients with AIP advocate caution.27 37 Gonadotropin-releasing hormone analogues reduce the severity and frequency of attacks in 60–80% of the women studied.27 38 39 In patients requiring prolonged treatment, we recommend oestrogen supplements to avoid menopausal symptoms, and specifically a patch to avoid first-pass hepatic metabolism.27 40 The literature suggests that haem–arginate is safe in pregnancy.41
Prognosis
The prognosis for a porphyric attack is good, with rapid resolution of abdominal pain, autonomic symptoms, seizures and encephalopathy once the attack is aborted. The neuropathy resolves more slowly, with the extent and timescale of the recovery depending on the magnitude of the axonal damage. Recovery usually occurs over many months, but delayed diagnosis may lead to incomplete return of motor function. Repeated attacks may result in an accumulation of damage, producing fixed weakness and atrophy.
Fewer than 10% of patients with AIP develop recurrent attacks without an identifiable precipitating factor.1 The long-term prognosis in patients with recurrent attacks is not as good as in those with a one-off attack. Recognised complications include hypertension, renal insufficiency, chronic pain and depression.9 There seems to be an increased risk of hepatocellular carcinoma in both symptomatic and asymptomatic patients with AIP. We therefore recommend screening those patients at risk.9 33 The mechanism for the increased risk of hepatocellular carcinoma is not fully understood, although there are two main hypotheses. One suggests that delta-aminolevulinic acid itself is carcinogenic, with its pro-oxidant and genotoxic properties.41 42 Alternatively, haem may be protective, and its relative reduction increases the risk.41 43 Ongoing research into the potential use of viral vectors and gene therapy gives promise for future treatment.41
Key points
Acute intermittent porphyria (AIP) should be suspected in patients with a combination of abdominal pain, psychiatric disturbance and peripheral neuropathy with prominent proximal weakness and retained reflexes.
The first step in diagnosing AIP is a urine test to determine the porphobilinogen to creatinine ratio.
The cornerstone of the management of an acute porphyric attack is a supplementary intravenous haemin preparation.
The choice of antiepileptic medication requires very careful consideration to avoid hepatic enzyme inducers (eg, phenytoin), which risk further worsening of the underlying condition.
Prevention is better than cure; avoiding precipitating factors such as alcohol, hormone treatments, medications and starvation can significantly reduce the risk of attacks.
Acknowledgments
We are grateful to the patient for allowing us to report her case and thank Mike Badminton (National Porphyria Service) for helpful discussions.
References
Footnotes
Contributors ROM: first draft of manuscript and revision. GR and PS: revision of first draft. OB: overall responsibility for revisions, submission and resubmission.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Herbert Bonkovsky, North Carolina, USA.
Other content recommended for you
- Unexpected presentation of acute porphyria
- Diagnosis and management of porphyria
- Update on the diagnosis and management of the autosomal dominant acute hepatic porphyrias
- Cerebral vasospasm and anterior circulation stroke secondary to an exacerbation of hereditary corproporphyria
- Neuropsychiatric porphyria in patients with refractory epilepsy: report of three cases
- A review of the clinical presentation, natural history and inheritance of variegate porphyria: its implausibility as the source of the ‘Royal Malady’
- Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow-up
- Urinary excretion of porphyrins, porphobilinogen and δ-aminolaevulinic acid following an attack of acute intermittent porphyria
- Porphyria for the neurologist: the bare essentials
- Acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome (PRES) and abdominal pain