Article Text

Abstract
Systemic amyloidosis can be hereditary or acquired. The autosomal dominant hereditary transthyretin amyloidosis and the acquired light-chain amyloidosis, the result of a plasma cell dyscrasia, are multisystem disorders with cardiovascular, autonomic and peripheral nerve involvement. There are numerous investigational modalities available to diagnose systemic amyloidosis and to assess the extent of organ involvement, but it is frequently misdiagnosed due to its heterogeneous clinical presentations and misleading investigation findings. An accurate and timely diagnosis of amyloid neuropathy can greatly impact on the outcomes for patients, especially as there will soon be new gene-silencing treatments for hereditary transthyretin amyloidosis.
- amyloid
- neuropathy
Statistics from Altmetric.com
Introduction
The amyloidoses are a rare group of diseases that result from extracellular deposition of amyloidogenic proteins (table 1).
Summary of the common types of amyloid and most frequently affected organs1 14 16 36
The hereditary amyloidoses, of which transthyretin (hATTR) is the most prevalent, are rare autosomal dominant disorders characterised by varying severity of peripheral and/or autonomic neuropathy, and other systemic manifestations, particularly cardiomyopathy. hATTR has been reported throughout the world, particularly in Europe, with marked phenotypic heterogeneity.1 TTR is primarily synthesised in the liver and misfolding of the ATTR protein causes aggregation and formation of insoluble amyloid fibrils that deposit systemically.2 Wild-type TTR is also a precursor of amyloid fibrils, usually resulting in a late-onset cardiomyopathy affecting approximately 25% of people over 80 years of age.3 Light chain (AL)-amyloidosis results from a plasma cell dyscrasia in which monoclonal plasma cells produce immunoglobulin light chain fragments that abnormally fold and deposit.4 AL-amyloidosis may be associated with myeloma or other B-cell malignancies, but most commonly the underlying haematological diagnosis is a benign monoclonal gammopathy of undetermined significance (MGUS). Lambda light chains are four times more commonly associated with AL-amyloidosis than kappa.5
In this review, we focus on hATTR and AL-amyloidosis as they are most commonly associated with a neuropathy. Recognising amyloid neuropathy is very important in this era of imminently available genetic therapies for hATTR, and improving therapies with better survival rates for patients with AL-amyloidosis.6
Major clinical features
hATTR and AL-amyloidosis have overlapping clinical manifestations, and both may present initially with isolated carpal tunnel syndrome. Patients then generally develop a peripheral and autonomic neuropathy and frequently cardiac involvement. Carpal tunnel syndrome can be the only symptom in hATTR in up to 33% of patients for a mean period of 4–6 years7 before other organs become clinically involved, and in AL-amyloidosis, it can predate other symptoms by over a year.8 The most common hATTR mutation worldwide is the V30M mutation that is endemic in Portugal, Japan and Sweden, but there are now over 100 known pathogenic ATTR mutations. In Portugal, V30M classically presents in the second or third decade with a painful or uncomfortable peripheral neuropathy with most patients describing paraesthesia or neuropathic pain initially rather than numbness progressing in a length-dependent pattern. Autonomic involvement can also be an initial symptom. In the UK, most patients have the T60A point mutation, which originally arose in north-west Ireland at least 200 years ago, and is widely prevalent in areas of high Irish immigration.9 Classically, there is early cardiac involvement, which can be the presenting symptom and usually occurs around 60 years of age. Symptoms and signs of peripheral neuropathy are the initial complaint in about half of patients, and often characterised by numbness rather than a painful neuropathy.10
As TTR is also produced within the choroid plexus and the retinal epithelium, central nervous system (CNS) manifestations can also occasionally occur in clinical practice, either from ongoing CNS deposition following liver transplant for V30M mutation or from hATTR mutations that have a predilection for the CNS. Oculoleptomeningeal amyloidosis, associated with the L12P mutation and other mutations can present with various neurological problems including seizures, subarachnoid haemorrhage, hearing or visual loss and headaches.11 The 10-year survival of patients with hATTR-V30M from liver transplant is 75%, and focal neurological episodes occur a median of 11 years after transplant. These include ischaemic and haemorrhagic strokes, short-lived, stereotyped episodes suggesting seizures or cortical transient ischaemic attacks. They have been difficult to classify as most patients have normal EEGs and CT scans, and MRIs are often contraindicated due to a pacemaker or implantable cardioverter defibrillator. However, histopathological studies of post-transplant brains show extensive cerebral amyloid angiopathy, negative anti-Aβ immunostaining, thereby suggesting ATTR deposition.12
Peripheral neuropathy
Neuropathic symptoms can be the presenting feature in up to 15% of patients with AL-amyloidosis.8 In both AL-amyloidosis and hATTR, patients typically describe a painful, length-dependent neuropathy starting in the feet, with numbness, burning and allodynia, which can be particularly troublesome at night. However, this can vary in hATTR depending on the mutation, such that patients with the V30M mutation from the endemic areas in Portugal present with this classic phenotype usually in the second or third decade, whereas patients with the T60A Irish mutation present usually in the fifth or sixth decade, and only 42% have a painful neuropathy. Presentation of hATTR in non-endemic areas, including patients with late onset V30M and many other mutations particularly if late onset, can be much more variable with a neuropathy characterised by the early involvement of all sensory fibres, and in some cases, more rapid progression.13 In the classic presentation, examination at an early stage may show only features of small fibre involvement with abnormalities in pinprick sensation and clinical features of median neuropathy at the wrists. Within months to years, large sensory fibres and motor nerve fibres become involved, resulting in impaired vibration and proprioception, and with weakness starting distally but progressing proximally requiring walking aids and eventually wheelchair use, and impaired upper limb function.14 Less common are focal cranial neuropathies, or plexopathies from focal deposition.14 Isolated amyloid myopathy or myopathy associated with systemic amyloidosis, either genetic or acquired, may also occur, with or without a neuropathy.15 16
Autonomic neuropathy
Up to 75% of patients with hATTR and 65% of patients with AL-amyloidosis develop symptoms of an autonomic neuropathy, affecting the cardiac, gastrointestinal and genitourinary systems.17 18 In our experience, diarrhoea and postural hypotension are among the most disabling symptoms of systemic amyloidosis. Orthostatic hypotension can be asymptomatic, or can cause persistent fatigue, light-headedness on standing or syncope. Gastrointestinal manifestations include gastroparesis, weight loss from early satiety and unexpected diarrhoea that may be socially prohibitive, often nocturnal especially initially, and may cause incontinence. Erectile dysfunction may be an early feature in men, and there may also be urinary frequency and retention. Pupillary and sweating abnormalities occasionally occur.14
Extraneurological manifestations
Amyloidosis can affect any organ or space, causing either dysfunction or organomegaly, for example hepatomegaly from AL-amyloid. Certain patterns of involvement strongly suggest amyloidosis, such as macroglossia and bilateral periorbital bruising, (‘racoon eyes’) for AL-amyloidosis. Nephritic or nephrotic syndrome can occur with both genetic and acquired forms.7 In addition, vitreous deposits occur in about 10% of patients with hATTR (never described with the Irish T60A mutation), sometimes visible on direct ophthalmoscopy. Significant cachexia in both hATTR and AL-amyloid is common, with patients easily losing more than 10% of body weight.
Symptoms of cardiac amyloidosis can present from either restrictive cardiomyopathy or arrhythmias. The cardiomyopathy causes symptoms of heart failure, which can be difficult to manage, requiring careful consideration of the patient’s fluid status and hypotension from autonomic neuropathy. Symptoms of arrhythmias such as syncope, reduced exercise tolerance and fatigue can also be difficult to distinguish from symptoms of orthostatic hypotension. Typical findings on echocardiography include thickening of ventricular walls; however, cardiac MR imaging is more sensitive.19
The diagnosis of amyloidosis is easier to recognise when patients present with a classic phenotype of symptomatic cardiomyopathy, previous bilateral carpal tunnel release and a progressive, length-dependent, painful sensorimotor neuropathy (figure 1). However, the neuropathy may be the only presenting feature and not be painful, and actively searching for other organ involvement can help to strengthen the case for investigating for amyloidosis.

Clinical manifestations of amyloidosis. AL, amyloid light chain. ICD, implantable cardioverter defibrillator
Approach to diagnosis
Confirm neuropathy
Patients are likely to present to neurologists with symptoms suggesting a peripheral neuropathy or be referred with a known diagnosis of amyloidosis to be investigated for a clinical or subclinical neuropathy. Standard nerve conduction studies can be normal in early small-fibre neuropathy. Testing for small fibre dysfunction depends on the locally available resources. Quantitative sensory testing is often used in assessment for small-fibre neuropathy and includes thermal detection and pain thresholds. The most validated technique to diagnose small-fibre neuropathy is quantification of intraepidermal nerve fibre density on a skin punch biopsy. In studies where small-fibre neuropathy was clinically suspected, this assessment had a sensitivity of 90% and specificity of 95%.20 With time, the large fibres are involved and classically, nerve conduction studies show a sensory more than motor, lower limb predominant axonal neuropathy with median nerve entrapment at the wrists.21 However, in both hATTR and AL amyloidosis, there can be slow conduction with prolonged distal motor latencies, which may lead to a neurophysiological diagnosis of a demyelinating neuropathy and subsequently a clinical diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). There is asymmetry in up to half of patients.
Investigate cause of neuropathy
Once a neuropathy is confirmed, the extent of investigations for the cause could be very minimal if there is a strong family history. Given the recent development of effective treatments for hATTR, we have a low threshold for requesting TTR gene sequencing in idiopathic axonal neuropathy or treatment resistant/atypical CIDP, especially if accompanied by carpal tunnel syndrome, autonomic or cardiac involvement, or the patient is of Irish ancestry. Testing is available on the UK National Health Service with results available within 4–6 weeks or sooner in an urgent situation. ATTR amyloidosis can be excluded if genetic testing for TTR is negative (no mutation), as sequence analysis of the gene detects over 99% of pathogenic variants.22 An online database (http://www.amyloidosismutations.com/main_menu.html) provides an updated list of amyloidogenic mutations and their phenotypes.5 Wild-type ATTR remains a possibility as it cannot be excluded on genetic testing.
If there is no family history, patients will usually undergo a broad, routine neuropathy screen looking for acquired causes. When considering AL-amyloidosis, it is important to identify the monoclonal plasma cells through searching for a paraprotein as well as the culpable light chain causing the amyloidosis. In patients with AL-amyloidosis, the sensitivity of serum protein electrophoresis for detecting a monoclonal protein is 66%, but this increases to over 90% if one combines serum electrophoresis with immunofixation and Bence Jones protein testing on urine. Serum-free light chain assay has a sensitivity of 88%.23 Lambda is the causative amyloid light chain four times more often than kappa light chains.5 In patients with renal impairment, the absolute values and the ratio need to be interpreted with caution. Patients with an identified abnormal light chain or paraprotein need referral to a haematologist.
Amyloidosis is a histological diagnosis. Given its multisystem involvement, in cases of an appropriate neurological phenotype, with no other obvious cause, and identification of a TTR mutation on genetic testing, or haematological findings suspicious for AL-amyloidosis, we do not necessarily pursue a nerve biopsy and sometimes use less invasive tissues to biopsy to achieve a diagnosis, such as an abdominal fat biopsy. However, in the absence of strongly supportive clinical findings, we do a nerve biopsy to search for amyloid and exclude other treatable causes. Identifying amyloid on sural nerve biopsies has a cited sensitivity as high as 86%; however, in real life, it can be challenging in an individual patient, given the patchy and sometimes proximal nature of amyloid deposition. It is not uncommon to identify only axonal degeneration in a nerve biopsy without identifying amyloid deposits, despite meticulous pathological examination.24
In the UK, once a diagnosis of amyloidosis is confirmed or strongly suspected, these patients are usually referred to the National Amyloidosis Centre to assess the extent of systemic involvement. If there is no previous histological confirmation of amyloidosis, patients usually undergo an abdominal fat aspiration, a simple and safe bedside procedure. The specificity of this test in identifying systemic amyloidosis approaches 100% with sensitivity varying between 52% and 88%.21 Congo red-positive areas of the formalin-fixed paraffin-embedded biopsy undergo immunohistochemistry for typing of the amyloid protein (TTR vs AL vs other), and if this is equivocal, the biopsy undergoes laser microdissection followed by mass spectrometry.25 These tests to identify protein type can be performed on any affected tissue, including salivary gland, nerve, rectal mucosa, endomyocardial biopsy specimens and tenosynovial tissues obtained at carpal tunnel release surgery.
The National Amyloidosis Centre and other specialist centres also have specialised imaging modalities to identify the degree of systemic amyloidosis and the pattern of uptake can provide clues to type of amyloid. Serum amyloid P (SAP) is a glycoprotein found in all types of amyloid deposits. SAP scintigraphy uses radiolabelled SAP as a tracer to quantify and identify amyloid deposition; however, heart, peripheral nerve and the CNS are poorly visualised. SAP scintigraphy has high sensitivity, 90%, in AA and AL amyloidosis,26 but only 48% in ATTR amyloidosis27 (figure 2). Another radionuclide tracer used is 99m-technetium-3,3,-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD). It is particularly sensitive and specific in identifying cardiac ATTR amyloid deposits, and ATTR deposits have different uptake and much higher sensitivity than AL-amyloid deposits26 (figure 3).

SAP scintigraphy in AL-amyloidosis compared with hATTR (posterior view). AL, amyloid light chain; hATTR, hereditary amyloid transthyretin; SAP, serum amyloid P.

(99m-technetium-3,3,-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD)). Practicalmyocardialscintigraphy in hATTR. hATTR, hereditary amyloid transthyretin.
MGUS may coexist with wild-type or hATTR, especially in older people, but this not widely appreciated. In a study of 57 patients with V122I mutation-related hATTR, aged 50–90 years (median 71 years), 49% had abnormal serum-free light chain ratios and/or paraprotein on immunofixation,28 suggesting neurologists may see abnormal haematological investigations commonly in these patients. Therefore, even when there are hATTR mutations, AL-amyloidosis is possible and vice versa. Hence, biopsies from different sites and organs may be necessary, and typing the amyloid fibrils is essential when there is concurrent ATTR mutation and a paraprotein.
Misdiagnoses/differentials
CIDP is the most common misdiagnosis of amyloidosis. This is plausible given patients with hATTR or AL-amyloidosis can have significant motor conduction velocity slowing. In one study, seven misdiagnosed patients fulfilled European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) criteria for definite demyelinating polyneuropathy (see clinical case 1). There were reduced conduction velocities as low as 33 m/s in the upper limbs and 30 m/s in the lower limbs.29 In a study of 150 patients with hATTR, 32% had been misdiagnosed, 61% of these were initially diagnosed as CIDP, and 2% as vasculitic neuropathy, which can also present as a painful, axonal neuropathy.29 Contributing to the risk of misdiagnosis is raised cerebrospinal fluid (CSF) protein in patients with systemic amyloidosis, and absence of clues to autonomic and systemic involvement at initial presentation. Given the frequent misdiagnosis, there should be high suspicion for hATTR in patients diagnosed with CIDP that do not respond to immunomodulatory treatment30 (other pitfalls are listed in Box 1).
Pitfalls in diagnosing amyloid neuropathy
History
No family history.
Minimal pain at onset.
Paraesthesia rather than pain.
Asymmetric.
No cardiac or autonomic symptoms.
Examination
Cranial nerve involvement.
Proximal weakness early.
Investigations
Concurrent paraprotein with hereditary amyloid transthyretin.
Raised CSF protein.
Slow conduction velocities on nerve conduction studies (NCS).
Myopathic units on Electromyography (EMG).
Failure to identify amyloid on first tissue biopsy may need repeat and varied tissues biopsied to confirm diagnosis.
Treatment
For AL-amyloidosis, high dose melphalan and autologous stem cell transplantation (HDM-ASCT) is the preferred first-line treatment for patients aged up to 65–70 years and eligibility depends on the extent of renal, cardiac, autonomic and bone marrow involvement. HDM-ASCT, in a carefully selected patient population, is associated with the best progression-free survival and overall survival, with treatment related mortality of 4%–13%.31 Patients not eligible for HDM-ASCT generally receive systemic chemotherapy with melphalan and dexamethasone or cyclophosphamide–bortezomib–dexamethasone (VCD).32 Prognosis depends on the extent of cardiac involvement (monitored through serum biomarkers) and the difference in quantities (rather than ratio) between involved (amyloidogenic light chain) and uninvolved serum-free light chain levels.
It is an exciting time for patients and clinicians in the field of hATTR. Historically, liver transplant was the first treatment option for hATTR, first performed in 1990 and while it increases survival in early onset ATTR-V30M (patients younger than 50 years), outcomes in non-V30M and older V30M patients are not as good. In recent times, TTR stabilisers, tafamidis and diflunisal have been used to treat patients with hATTR. These drugs bind to thyroxine-binding sites of TTR and inhibit TTR tetramer dissociation (an essential step for amyloid formation), and there is some evidence that they may slow disease progression, especially in the V30M patient population.33 This year, there has been significant progress made in treatment for hATTR with two types of gene-silencing therapies, inotersen, an antisense oligonucleotides and patisiran, a small interfering RNAs, achieving their primary outcome measures in two separate randomised placebo-controlled, phase 3 trials.34 35 Inotersen is a subcutaneous preparation and is administered weekly, while patisiran is administered intravenously, 3 weekly. They both reduce the production of mutant and wild-type TTR through different mechanisms (for further information on mechanisms, please read6). These therapies are a welcome addition to the treatment options for patients with V30M and non-V30M mutations.
Conclusion
In this era of rapidly evolving gene therapies for hATTR and improving therapies for AL-amyloidosis, an accurate and timely diagnosis of amyloid neuropathy can greatly impact outcomes for patients. There is considerable heterogeneity in the clinical presentation of amyloid neuropathy but having a high clinical suspicion together with knowledge of the advantages and limitations of the various diagnostic techniques used is key to making a diagnosis.
Case 1 (TTR misdiagnosed as CIDP)
A 70-year-old woman of Northern Irish background, was initially diagnosed as having CIDP based on asymmetrical, distal, sensory-onset neuropathy, raised CSF protein of 0.69 g/L and prolonged distal motor latencies and slowed conduction velocities on neurophysiology. She was referred to our centre after failing to respond to prednisolone, intravenous immunoglobulin and plasma exchange. At age 66 years, she developed unilateral, uncomfortable paraesthesia in her left foot, which over a year spread to both knees. Following this, she developed proximal weakness with trouble standing up from sitting which progressed such that she required a wheelchair 3 years after disease onset. There was no weight loss, early satiety, bowel or bladder symptoms but she did have postural dizziness, which developed 2 years in to her illness. Her other history included atrial fibrillation, diagnosed at age of 68 years. There was no family history of neurological disease.
On examination, 4 years after the onset of her illness, she had upper limb weakness of MRC 4 proximally and 0–1 distally. On lower limb examination, there was pitting oedema to her ankles, global weakness with proximal power of MRC 2–3 and distal power of 0–2. She was areflexic and had downgoing plantars. Sensory examination identified reduced pinprick to her knees and elbows, normal joint-position sense and reduced vibration to ankles.
Neurophysiology showed a mixed, demyelinating and axonal neuropathy (table 2).
Case 1 neurophysiology
CSF analysis showed a raised protein of 0.69 g/L. Sural nerve biopsy identified endoneurial deposits that stained strongly with Congo red and displayed apple-green birefringence when viewed under polarised light (figure 4). Immunotyping suggested transthyretin type of amyloid. Sequencing of the TTR gene revealed the T60A pathogenic mutation. DPD scan at the National Amyloidosis Centre showed moderate amount of cardiac amyloid. She was treated with diflusinal, a TTR stabiliser.

{kind=link}
{kind=link}
{kind=link}
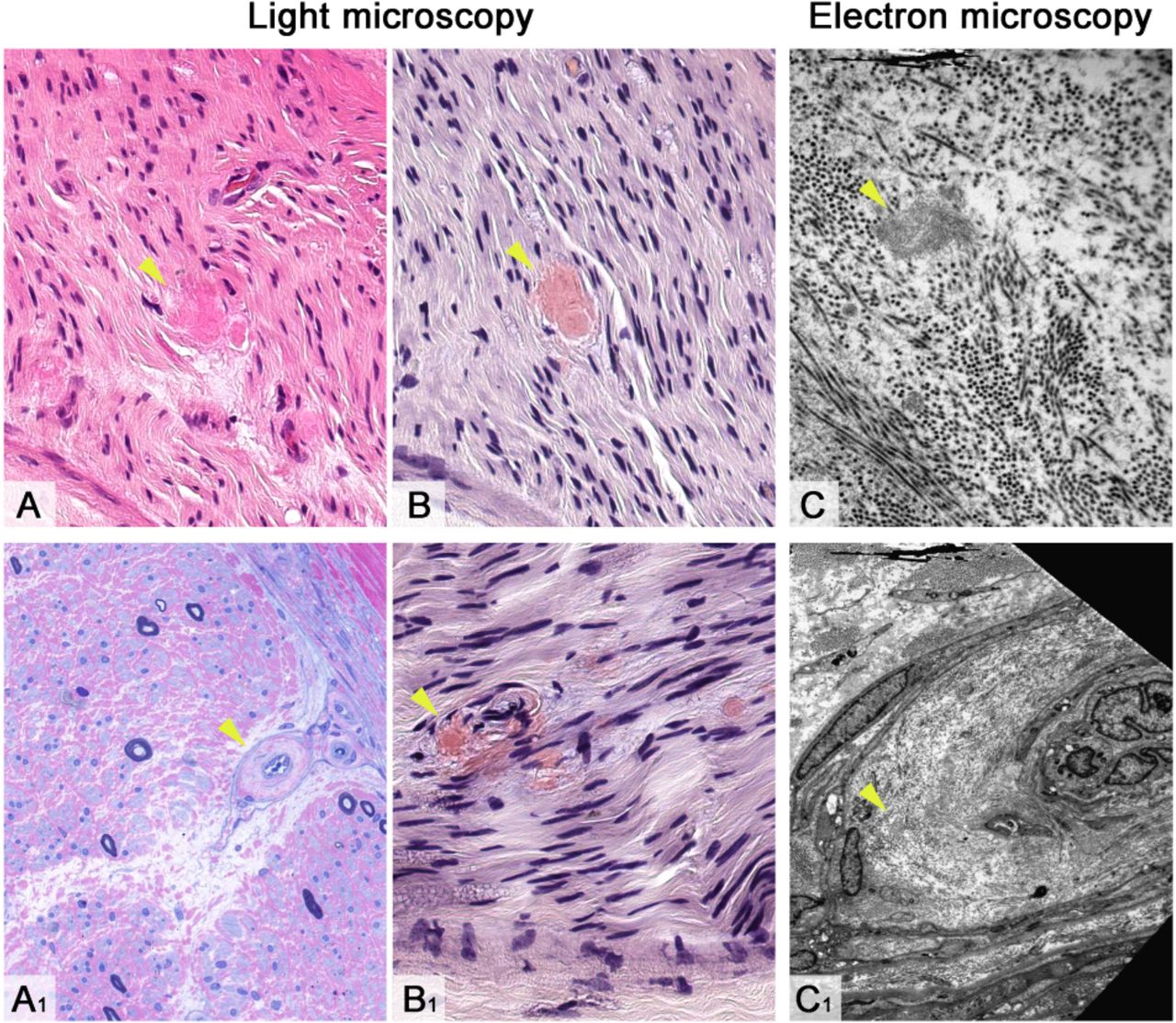{kind=link}
Morphological appearances of amyloid neuropathy due to amutation in the TTR gene (biopsy of sural nerve). Endoneural amyloid deposits on H&E stained section (A) are seen as amorphous brightly eosinophilic deposits. Semi-thin resinsection stained with methylene blue azure–basic fuchsin (A1) shows substantial loss of large and small myelinated fibres across the transversely oriented fascicle with no evidence of regeneration or active degeneration; amyloid deposition is evident in an endoneural blood vessel resulting in a concentrically thickened vessel wall. Congo red histochemical staining (B and B1) accentuates amyloid deposits in the endoneurium (B) and blood vessel wall (B1). Electron microscopy further highlights amyloid fibrils freelyin the endoneurium (C) and within a blood vessel wall (C1). Amyloid deposits in all images are highlighted with a yellow arrowhead. Scale bar: 40 μm in A, A1, B and B1; 5 μm in C and C1 .Images courtesy of Dr Zane Jaunmuktane.
Case 2 (AL difficult to diagnose on tissue)
A 70-year-old woman with a 6-year history of Waldenstrom’s macroglobulinaemia, previously treated with fludarabine and cyclophosphamide with partial haematological response secondary to early cessation of treatment due to side effects, presented with a 1-year history of shooting pains in the calves and burning in the soles of the feet. Three months before referral to our centre, she had developed weakness in her feet and difficulty in climbing stairs progressing to using a wheelchair after a further 3 months.
On initial examination, there was no postural hypotension. Cranial nerve examination was normal except for a left tonic pupil. In her upper limbs, there was bilateral wasting of the intrinsic hand muscles. Power was normal proximally and MRC 4 to 5 in the intrinsic hand muscles. Reflexes were present. In the lower limbs, she could not stand from a chair without using her hands and had bilateral wasting of extensor digitorum brevis. Tone was normal, and there was proximal and distal weakness, with hip flexors weak at MRC 4 and ankle dorsiflexion was 4 on the right and 2 on the left. Knee reflexes were present, ankle jerks were absent and plantars were unresponsive. Sensory examination showed pinprick abnormal to mid-thighs with no abnormality in the upper limbs, vibration was reduced to the costal margins and proprioception was normal.
Initial investigations showed an IgM lambda paraprotein of 5.1 g/L, normal serum-free light chains with a normal ratio and no urine Bence Jones protein. Neurophysiology revealed abnormal thermal thresholds in the lower limbs suggesting a small-fibre neuropathy with no large fibre involvement. Repeat neurophysiology 6 months later showed an axonal, asymmetric neuropathy (table 3).
Case 2 neurophysiology
She underwent a superficial peroneal and peroneus brevis biopsy that showed a moderate loss of large myelinated fibres and moderate loss of all axons. Immunohistochemical studies showed endoneurial labelling for lambda light chains, however, Congo red staining at our institution was negative. The biopsy was sent to National Amyloidosis Centre for further studies, and using antibodies against SAP, they were able to show amyloid deposits present in the nerve bundles. Antibodies against SAP, which co-deposits with amyloid, can be used to distinguish light chain deposits of AL amyloidosis from monoclonal immunoglobulin deposition disease (19). Assessment at the National Amyloidosis Centre identified nephritic range proteinuria and SAP scintigraphy showed moderate splenic amyloid load as well as equivocal liver involvement, with no evidence of cardiac amyloidosis.
Her mobility continued to deteriorate, and she developed features of autonomic neuropathy with urinary retention, diarrhoea and orthostatic hypotension. She was treated with cyclophosphamide, dexamethasone and rituximab and achieved sustained partial remission.
Systemic amyloidosis can be hereditary or acquired with hereditary transthyretin (TTR) amyloidosis being the most common inherited form.
Systemic amyloidosis is a multi-system disorder with varying involvement of the peripheral and autonomic nerves, and particularly, cardiac involvement.
The classic neuropathic presentation is of a painful, length dependent neuropathy but atypical presentations without pain or presentations of focal neuropathies, plexopathies or myopathy, are not uncommon.
Classically, neurophysiology suggests an axonal neuropathy, but standard nerve conduction studies can be normal in early small-fibre neuropathy, and slow conduction velocities or prolonged distal motor latencies can also be seen leading to a clinical misdiagnosis of CIDP.
Older patient may have concurrent hereditary TTR amyloidosis and a paraprotein which can complicate identifying AL vs. TTR amyloidosis.
Consider requesting TTR gene sequencing in patients diagnosed with CIDP who do not respond to immunomodulatory treatment.
There is exciting progress being made in treating hereditary TTR amyloidosis with 2 gene-silencing therapies showing promising results in separate randomised, placebo-controlled, phase 3 trials.
References
Footnotes
Contributors MMR: designed the study. MK: collected data and wrote a draft of the paper. All other authors provided detailed written edits and multiple further drafts of the review for publication. All authors work fulfilled the following: substantial contributions to the conception or design of the work or the acquisition, analysis or interpretation of data; drafting the work or revising it critically for important intellectual content; gave final approval of the version published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding AMR is funded by a Wellcome Trust Postdoctoral Fellowship for Clinicians (110043/Z/15/Z). MMR is grateful to the Medical Research Council (MRC), MRC Centre grant (G0601943) and the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) for their support. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. We are also grateful to David Hutt from National Amyloid Centre for providing the images from investigations.
Competing interests AMR has received support from Alnylam UK Limited to attend scientific meetings and an honorarium for speaking at a sponsored symposium.
Patient consent for publication Obtained.
Provenance and peer review Commissioned. Externally peer reviewed by Davide Pareyson, Milan, Italy.
Other content recommended for you
- Cardiac transthyretin amyloidosis
- The transthyretin amyloidoses: advances in therapy
- Light-chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response
- Amyloid in the cardiovascular system: a review
- A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK
- Amyloidosis
- Amyloid diseases of the heart: assessment, diagnosis, and referral
- Diagnosis and management of sensory polyneuropathy
- Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy
- A rare cause of weakness