Article Text

Abstract
Progressive myelopathy is a rare complication of chronic hepatic disease which has never been reported in the paediatric age group. We describe the 11 year course of an adolescent male with hepatic myelopathy caused by cryptogenic micronodular cirrhosis. His condition has been associated with persistent polycythaemia and extraordinary increases of whole blood manganese, with magnetic resonance imaging evidence of manganese deposition within the basal ganglia and other regions of the brain. The patient has developed neither liver failure nor parkinsonism. The pathophysiological bases of this multiorgan system disorder are described.
- hepatic myelopathy
- manganese
- neurotoxicity
- erythropoietin
Statistics from Altmetric.com
Hepatic myelopathy is a rare neurological complication of chronic liver disease that has only been reported in adults.1-6 Affected patients develop a disabling progressive spastic paraparesis which is almost always associated with overt liver failure and a systemic portal–caval shunt. While hepatic myelopathy has been reported in patients with hepatic failure secondary to congenital liver disorders, these affected individuals did not develop neurological symptoms until adulthood.1 ,3 We report the first case of hepatic myelopathy associated with cirrhosis presenting during adolescence. Additional unusual features of this patient's disorder are chronic polycythaemia and extraordinary increases of whole blood manganese, together with magnetic resonance (MR) imaging evidence of heavy metal deposition in the basal ganglia. Despite these features, throughout 11 years of observation, the patient has not developed extrapyramidal symptoms or clinical signs of decompensated cirrhosis.
Case report
A 14 year old white male presented for evaluation of malaise, and stiffness of gait of two months duration. Abnormal laboratory studies included a haematocrit of 0.67, a haemoglobin of 2.25 g/l, and a mean corpuscular volume of 79.3 fL. Mild hepatomegaly and increased tendon reflexes were noted, and there was no evidence of malignancy, haemoglobinopathy, abnormal oxygen–haemoglobin dissociation, or increased erythropoietin. There was no evidence of cardiopulmonary disease and his arterial oxygen saturation was normal. His past history was significant for three hospitalisations between 5 and 12 months of age for treatment of aseptic meningitis. With the exception of mild language delay, his early childhood development was normal. There was no family history of consanguinity or of neurological, hepatic, or haematological disease. His polycythaemia was treated successfully with biweekly phlebotomies.
Over the next year, he developed progressive leg weakness and stiffness of gait. Neurological examination revealed a spastic paraparesis with normal upper extremity function, no sphincter involvement, normal cranial nerve function, and a mild sensory loss in the distal extremities to pinprick and cold temperature without evidence of a sensory level. There was no evidence of cerebellar or extrapyramidal dysfunction.
Laboratory evaluations included normal measurements of serum lead, free red blood cell protoporphyrin, vitamin E, vitamin B12, folate, erythrocyte sedimentation rate, C reactive protein, very long chain fatty acids, and urine amino acids. He had a normal karyotype, and a urine metabolic screen was negative as were antinuclear antibody, rheumatoid factor, rapid plasmin reagin, and serology for HTLV-I andBorrelia burgdorferi. Spinal fluid analysis was unremarkable with a protein of 0.24 g/l, normal cytology, and no evidence of oligoclonal bands.
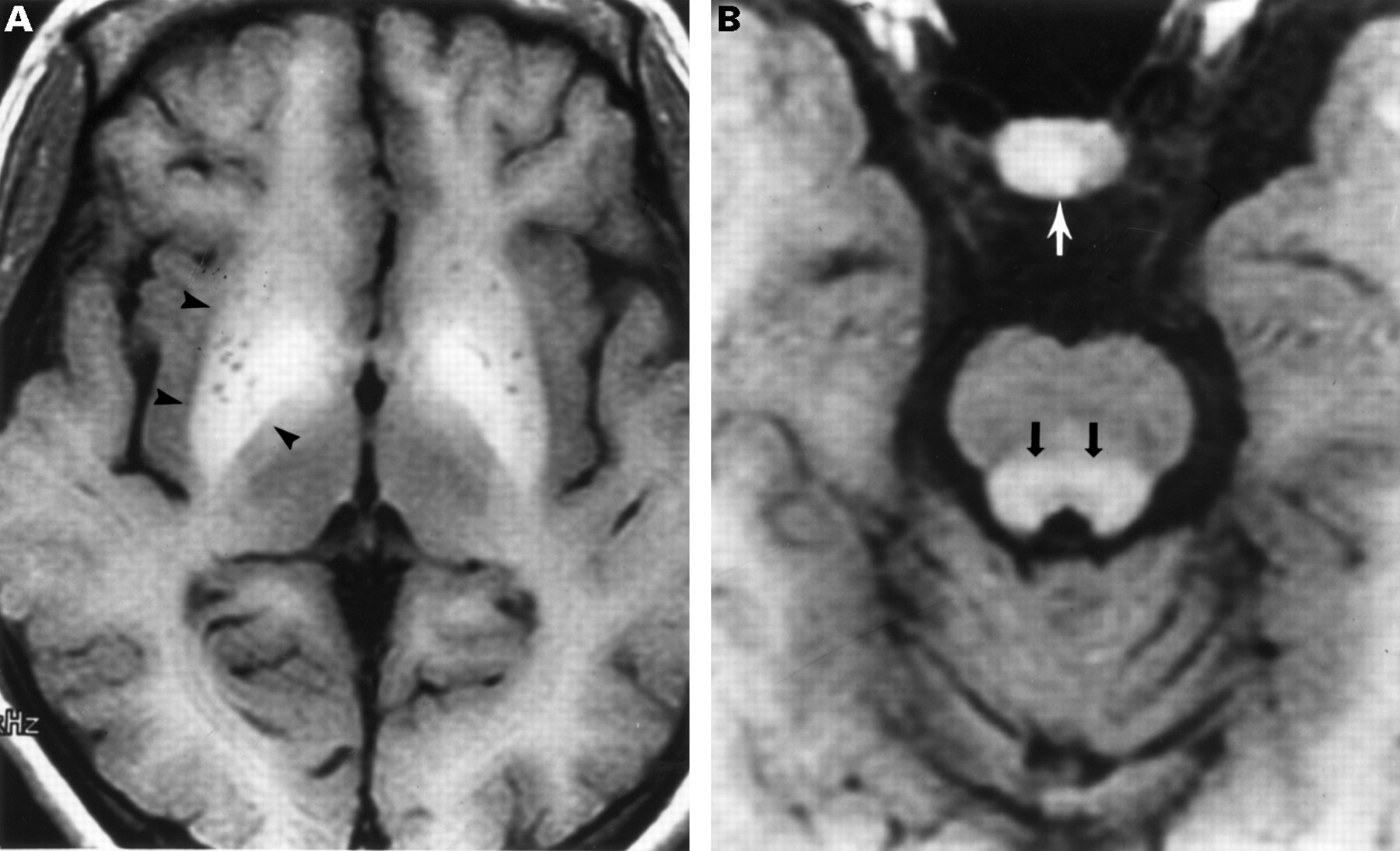
MR imaging of the spinal cord was normal. However, sagittal and axial T1 weighted cranial MR imaging showed high signal in the lenticulate and dentate nuclei, brainstem, and pituitary, consistent with the deposition of manganese7-10; brainstem atrophy was also present (fig 1). T2 weighted MR imaging was normal. Whole blood manganese, measured via flameless atomic absorption spectrophotometry,11 was greatly increased at 3.48 μmol/l (normal 0.15 (SD 0.05)).
{kind=link}
Magnetic resonance imaging of the brain performed at age 19. (A) Axial T1 weighted image shows increased signal in the lenticulate nuclei (arrowheads), greatest in the globus pallidi. Tiny low signal areas within the nuclei represent perivascular spaces of the lenticulostriate arteries. (B) Axial T1 weighted image shows high signal in the pituitary gland (white arrow) and in the brainstem (black arrows). Brainstem atrophy is present.
In order to determine whether the patient's increased whole blood manganese concentration was due, in part, to either environmental exposure or a high consumption of manganese rich containing food or supplements, a home visit was made. A three day food recall did not reveal an excessive ingestion of manganese. The patient lived in a residential section of a small Sacramento Valley, California town, and there were no industrial or dump sites nearby. An analysis of the tap water from both the current and previous residences of the patient did not reveal increased concentrations of manganese. Whole blood manganese concentrations of the patient's half sister and neighbour were normal at 0.22 μmol/l and 0.11 μmol/l, respectively. Interestingly, the mother's concentration was increased at 0.38 μmol/l.
To determine if there was evidence of heavy metal deposition in other organs, MR imaging of the chest, abdomen, and pelvis was performed. This study revealed hepatosplenomegaly with a slight diffuse increase in T1 weighted signal in the liver. A liver biopsy revealed changes consistent with a diagnosis of micronodular cirrhosis including distortion of the architecture by micronodules, focal liver cell dropout, occasional binucleated cells, abundant granular cytoplasm, and no evidence of steatosis or of increased iron stores. There was no indication of hepatic copper deposition via rubeanic acid and rhodamine staining. There was no immunohistochemical evidence of intrahepatic erythropoietin.12 Liver manganese concentration, measured via flame atomic absorption,13 was increased at 0.15 μmol/g (normal 0.05 (SD 0.03)).
The patient had no previous history of jaundice or of exposure to hepatitis, and no alcohol or hepatotoxic drug use. Serological and virological studies for hepatitis B and C viruses and cytomegalovirus were negative, while serological studies for Epstein–Barr virus were consistent with past infection. The patient had no abnormalities of hepatic transaminase activities, bilirubin, ammonia, coagulation, α1 antitrypsin, or ceruloplasmin. Serum iron was low at 3.7 μmol/l, iron binding capacity was increased at 116 μmol/l, and ferritin was reduced at 6 μg/l; therefore, there was no evidence of haemachromatosis. The patient does not have Kayser–Fleischer rings, and his urine copper:creatinine ratio of 0.049 is within the normal range (0.013–0.071, GJ Brewer, unpublished data), making a diagnosis of Wilson's disease very unlikely. Thus, the patient has a diagnosis of cryptogenic micronodular cirrhosis. A hepatic ultrasound revealed hepatomegaly and no increased echogenicity, and a Doppler flow study showed normal hepatic blood flow without evidence of a portal–caval shunt, portal collaterals, or varices. There was no evidence of oesophageal varices on oesophagoscopy.
During 11 years of clinical observation, the patient has had progressive spastic weakness of the legs leading to loss of ambulation and painful extensor spasms which are being treated with intrathecal baclofen. The changes documented on MR imaging of the brain have been stable, and an electroencephalogram (EEG) shows slight diffuse slowing. He has developed neither mental status changes nor extrapyramidal dysfunction. Whole blood manganese concentrations have consistently remained increased, ranging from 2.93 to 3.66 μmol/l. The patient has had persistent polycythaemia, requiring twice weekly phlebotomy. During this time period, erythropoietin concentrations have ranged from 5 to 473 U/l (normal 0–16 U/l) and these concentrations have not correlated with the haematocrit. The patient has had no deterioration of hepatic function, plasma ammonia is 30 μmol/l, serum α fetoprotein is 10.7 μg/l, and there has been no radiographic evidence of the development of a hepatic malignancy. While he has a firm, irregular, normal sized liver, he has no stigmata of chronic liver disease such as jaundice, palmar erythema, spider nevi, prominent abdominal veins, or ascites.
Discussion
This patient presented with polycythaemia together with a progressive spastic paraparesis. His initial evaluation did not reveal a cause for the erythrocytosis which has been controlled with twice weekly phlebotomy. His myelopathy did not have an infectious, inflammatory, demyelinating, dysmyelinating, or neoplastic aetiology. Insight into the pathogenesis of this boy's complex disorder was obtained when cranial MR imaging showed manganese deposition within the basal ganglia which then led to the diagnosis of a cryptogenic micronodular cirrhosis.
Hepatic myelopathy is a progressive irreversible disorder which leads to spastic weakness of the legs with minimal involvement of the upper extremities. Affected patients do not have bowel or bladder dysfunction or sensory disturbances, unless a significant neuropathy, usually caused by alcoholism, is also present. In the majority of cases the myelopathy develops after several episodes of hepatic encephalopathy,1 but rarely this neurological syndrome may be the presenting sign of liver failure.5 Almost universally, patients have evidence of either spontaneous or surgical portal–systemic shunting, and it has been hypothesised that ammonia or other metabolites bypassing the liver result in progressive neurotoxicity.1 ,3 ,4 Neuropathological studies have shown demyelination of the corticospinal tracts with variable axonal loss. These lesions are primarily within the cervical levels of the spinal cord and occasionally within the brainstem, with little to no involvement of other tracts.1 ,4 ,5
Spinal cord MR imaging has been performed in four previously reported cases of hepatic myelopathy1 ,2 ,4 ,5; as in our case, the imaging was normal. Cranial MR imaging was reported in three of these cases; none of them had brainstem atrophy while one had evidence of heavy metal deposition within the basal ganglia.1 These deposits, typically thought to be rich in manganese, are common in individuals with chronic liver disease of diverse aetiologies; frequently these patients have extrapyramidal dysfunction, which was not present in our patient.7-10 A variety of clinical studies have shown increased blood manganese concentrations in these patients,7-9 and studies of postmortem specimens have shown increased tissue manganese concentrations within the basal ganglia.9 ,10 ,14 Whole blood manganese concentrations in affected patients have been reported to range from 0.27 to 2.02 μmol/l,7-9 values which are lower than the concentrations measured in our patient (2.93–3.66 μmol/l). However, his blood manganese concentrations were in the range measured in individuals with parkinsonism secondary to chronic industrial manganese intoxication (1.84–7.38 μmol/l).15 While the patient's mother had a higher than normal concentration of blood manganese, her manganese absorption values were only slightly increased16(data not shown), and she had normal brain MR imaging.
It is estimated that 2–10% of dietary manganese is absorbed into the blood. Manganese in blood is primarily associated with erythrocytes, while manganese in the plasma fraction is primarily associated with transferrin and albumin. The liver effectively clears this metal, with greater than 90% of the absorbed manganese typically being excreted into the bile within 24 hours.17 Manganese clearance can be impaired in patients with portal–systemic shunts, cholestasis, and hepatocellular disease, which can lead to increased blood concentrations and manganese deposition within the basal ganglia and other regions of the brain.8-10 ,14 Elevated blood manganese concentrations and MR imaging evidence of basal ganglia heavy metal deposition has also been reported in patients receiving chronic parenteral nutrition with high manganese concentrations. Significantly, there was resolution of the MR imaging abnormalities after manganese was removed from the parenteral nutrition solution.18 ,19
While it could be speculated that the high whole blood manganese concentrations observed in this subject were secondary to a higher than normal amount of dietary manganese uptake, two lines of evidence argue against this. Firstly, the dietary history of the patient did not reveal a high amount of manganese intake. Secondly, although the transport of manganese across the intestinal tract is still poorly understood, it is thought to occur through mechanisms similar to those regulating non-haem iron uptake.17 ,20 Our observation of low to normal iron stores in this patient would argue against a higher than normal intestinal uptake of these two metals. Given the above, we suggest that the manganese accumulation in this individual is caused by an impairment in the secretion of excess manganese into the gut, rather than an increased absorption of the metal.
The current patient presented with polycythaemia, which has persisted for 11 years. Polycythaemia has been reported in cirrhotic patients, either caused by hepatic synthesis of erythropoietin from an associated hepatocellular carcinoma,12 or presumably caused by chronic hypoxia from intrapulmonary shunting or portal–pulmonary shunts.21 While our patient does have increased erythropoietin, he does not have evidence of either a hepatic tumour, or hepatic synthesis of erythropoietin. In addition, he has not shown any evidence of hypoxia, including cyanosis or clubbing. Alternatively, patients with cirrhosis may have abnormal hepatic clearance of erythropoietin, thereby leading to polycythaemia.22 This may be the cause of the polycythaemia in our patient. In addition, manganese and other transition metals have been shown in animal studies to have effects on erythropoietin gene regulation which are similar to those induced by hypoxia.23 Therefore, the increased manganese concentrations in our patient may also play a role in the pathogenesis of the polycythaemia.
While our patient is the youngest individual reported with hepatic myelopathy, there are other unique features of his disorder. With the exception of slight slowing of the EEG, which may indicate a mild hepatic encephalopathy, he has had no other evidence of either liver failure or portal–systemic shunting. Indeed, his cirrhosis was cryptogenic, and was diagnosed only after abdominal imaging showed hepatosplenomegaly and a subsequent liver biopsy. To our knowledge, there have been no other cases of hepatic myelopathy reported in patients without evidence of either liver failure or portal–systemic shunting. This patient also has extraordinary increases of whole blood manganese with manganese deposition within the basal ganglia and other brain regions. Despite these findings, presently he has no clinical evidence of extrapyramidal dysfunction. This patient's disorder is also characterised by increased liver manganese concentrations. It is interesting to speculate that the increased liver manganese may have contributed to either the initiation or progression of the cirrhosis. Acute loads of manganese have been shown to result in the induction of intrahepatic cholestasis.24 In addition, changes in cellular manganese concentration can give rise to alterations in the redox potential of the cell. The functional consequences of these changes are not yet understood.17
The pathophysiological mechanism of hepatic myelopathy in our patient has not been established. As many patients with chronic liver disease and increased blood manganese have extrapyramidal symptoms but no evidence of a myelopathy,7-10 we cannot firmly establish that our patient's hepatic myelopathy is a result of manganese neurotoxicity. Alternatively, some other neurotoxin which is not cleared by this patient's cirrhotic liver may have led to corticospinal tract demyelination. While this patient does not have clinical extrapyramidal dysfunction, given his extraordinary whole blood manganese concentrations, one would expect that he is at high risk of developing parkinsonism. Therefore, our patient may be a candidate for liver transplantation. In a patient with Alagille syndrome, liver transplantation has been shown to improve extrapyramidal symptoms and to reverse the basal ganglia changes shown by MR imaging.7 While hepatic myelopathy is considered to be irreversible, one of the two patients with this disorder who underwent liver transplantation showed some improvement in her spastic paraparesis and eventually could ambulate with a cane.2 ,6
Acknowledgments
The authors wish to thank Dr George J Brewer of the University of Michigan for his interpretation of the urinary copper excretion in this patient and for reviewing the manuscript. This work was supported, in part, by the Children's Miracle Network Research Program, University of California, Davis.