Article Text

Abstract
OBJECTIVE To investigate whether the stiff limb syndrome may be separated from the stiff man syndrome and progressive encephalomyelitis with rigidity on simple clinical grounds, and whether such a distinction has implications for aetiology, treatment, and prognosis.
METHODS Twenty three patients referred over a 10 year period with rigidity and spasms in association with continuous motor unit activity, but without evidence of neuromyotonia, extrapyramidal or pyramidal dysfunction or focal lesions of the spinal cord were reviewed. The patients were divided into those with an acute or subacute illness, leading to death within 1 year, and those with a chronic course. The latter were divided into those in whom rigidity and spasms dominated in the axial muscles, or in one or more distal limbs, at the time of their first assessment.
RESULTS This simple division identified three distinct groups of patients. (1) Progressive encephalomyelitis with rigidity: two patients had a rapidly progressive condition characterised by widespread rigidity which resulted in death within 6 and 16 weeks. One patient had negative anti-GAD and anti-neuronal antibodies, but had markedly abnormal CSF and widespread denervation. The principal pathological findings in this case were a subacute encephalomyelitis which primarily affected the grey matter. In the remaining patient anti-GAD antibodies were not tested, and postmortem was refused. (2) Stiff man syndrome: eight patients had rigidity and painful spasms of the lumbar paraspinal, abdominal, and occasionally proximal leg muscles associated with a lumbar hyperlordosis. There was no involvement of the upper limbs, distal lower limbs, sphincters or cranial nerves. Seven had anti-GAD antibodies and most had additional evidence of autoimmune disease. Neurophysiologically there was continuous motor unit activity with abnormal exteroceptive reflexes, but a normal interference pattern during spasms. The patients all responded to baclofen/diazepam and remained ambulant. (3) Stiff limb syndrome: thirteen patients had rigidity, painful spasm, and abnormal postures of the distal limb, usually the leg. About half went on to develop sphincter or brainstem involvement. Generalised myoclonic jerks were not a feature. Only two had truncal rigidity, and another two had anti-GAD antibodies. Most had no evidence of autoimmune disease. Neurophysiologically they had continuous motor unit activity in the affected limb, abnormal exteroceptive reflexes, and abnormally segmented EMG activity during spasms. The disease ran a protracted course, and most patients had only a partial response to baclofen or diazepam. About half became wheelchair bound.
CONCLUSIONS The stiff limb syndrome seems distinct from the stiff man syndrome or progressive encephalomyelitis with rigidity, and is an important cause of rigidity and spasm in the setting of continuous motor unit activity.
- stiff man syndrome
- progressive encephalomyelitis with rigidity
- stiff limb syndrome
Statistics from Altmetric.com
The combination of rigidity, reflex, and action induced spasms and continuous motor unit activity at rest is rare, and may be due to tetanus, encephalomyelitis, corticobasal degeneration, focal lesions of the spinal cord, or the stiff man syndrome. Stiff man syndrome is a chronic disorder involving stiffness and painful spasms of the trunk.1 An exaggerated lumbar lordosis is an almost universal finding, but signs of cerebral, brainstem, or long tract disturbance are absent. The diagnosis is supported by the finding of anti-GAD antibodies in blood and CSF. However, most series to date report the presence of anti-GAD antibodies in only around 60% of patients.2 3 Moreover, many patients do not show the classic axial distribution of stiffness and spasms. Recently, Brownet al reported a small series of patients in whom stiffness and spasms remained confined to the lower limbs.4 These patients were anti-GAD negative, and it was suggested that they represented a distinct syndrome, possibly due to a chronic spinal interneuronitis. Here, we review the 23 patients of stiff people who presented to our hospital over a 10 year period, in an attempt to redefine the conditions that may present with rigidity and spasm in association with continuous motor unit activity at rest.
Patients and methods
All patients referred to this hospital between 1986 and 1995 with a diagnosis of possible stiff man syndrome were analysed. Patients were examined by the authors and those with extrapyramidal disease, spasticity, corticobasal degeneration, dystonia, reflex sympathetic dystrophy, neuromyotonia, or structural lesions of the spinal cord were excluded. The 23 patients identified all had, at some point, evidence of continuous motor unit activity and were analysed in terms of their clinical features, investigations, response to therapy, and natural history. Six patients were no longer under follow up at this hospital, and in these patients their current clinical condition was assessed through contact with the patients’ general practitioner or local neurologist. Two patients died. Four of the patients with distal stiffness, and two of those with axial stiffness have been previously described.4-6
Anti-GAD antibodies were measured by immunoprecipitation of a radiolabelled recombinant human GAD 65 antigen. Details of the techniques used for surface polymyography, and the recording of cutaneomuscular and startle responses have been previously published.4 The response to therapy was assessed using the medical notes, as no standard objective measure of response was employed in all patients. Such a response was graded in terms of either (a) no response; (b) improvement in stiffness and spasms with only a transient improvement in mobility; and (c) a sustained improvement in mobility. Limited sample sizes precluded useful statistical analysis.
Results
A total of 23 patients were entered into the study for analysis. After review, it became apparent that these patients could be divided into two groups: those with progressive encephalomyelitis with rigidity, an acute or subacute illness leading to death within 1 year, and those with a chronic course. The second group were further divided into those in whom rigidity and spasms dominated in the axial muscles (of the neck, trunk, and proximal lower limb), and those in whom rigidity and spasms dominated in one or more distal limbs, by the time of their first assessment (3 months to 4 years after onset of the illness). In practice the distinction was relatively absolute, with only two out of the 13 patients with stiff limbs having any evidence of axial rigidity. In one of these patients axial difficulties had been delayed by several years and, in both they were overshadowed by distal stiffness and spasm by the time of their first assessment. Patients with axial rigidity met the criteria of Lorish et al for the diagnosis of the stiff man syndrome, with the exception of treatment response, which we do not consider an absolute prerequisite for the diagnosis.7 Those with distal rigidity had a condition similar to that described by Brown et al.4 We call this the stiff limb syndrome.
CLINICAL FEATURES
There were no distinct differences in the sex distribution or age of patients with either axial or distal rigidity (table 1). The duration of illness (1 to 19 years) was also little different between these two groups, although the patients with progressive encephalomyelitis with rigidity succumbed rapidly to his disorder, both dying within weeks of developing their first symptoms of rigidity.
Clinical features in stiff people
The presenting features of these different disorders were almost entirely in accord with the differential distribution of rigidity on examination. Thus seven out of eight patients with axial stiffness and the stiff man syndrome complained of stiffness and painful spasms of their lower back with some involvement of their proximal legs, and associated difficulty in walking. The initial symptom in one patient in this group was stiffness of the whole leg, but marked involvement of the lumbar paraspinal and abdominal musculature dominated the clinical picture by the time of his first assessment. By contrast, the initial complaint in patients with distal rigidity (stiff limb syndrome) was stiffness and painful spasm of a limb, usually of the calf and foot, with difficulty walking. Spasms in all groups could be spontaneous, reflex, or provoked by voluntary action. Although spasms could have a jerky quality, particularly in those with distal rigidity, none of the patients had generalised myoclonic jerks.
All but one patient with axial rigidity had an exaggerated lumbar lordosis, but none had any fixed abnormal posturing of a distal limb. By contrast, the latter was a universal finding among those with distal rigidity. In two patients the distal arm rather than the leg was involved in this group. In the one patient with distal rigidity but early symptomatic involvement of the truncal musculature, this was not associated with a lumbar hyperlordosis until 3–4 years after the lower limb had become clinically involved. In the other case of involvement of the truncal musculature with distal rigidity, these symptoms developed 16 years after the onset of the disease. Progressive encephalomyelitis with rigidity began with bulbar involvement. Within days to weeks the limbs, then the truncal muscles were affected.
The natural history was different in the three groups. Patients with axial stiffness (stiff man syndrome) seemed to progress and then stabilise after months to years. By contrast, seven patients with distal stiffness (stiff limb syndrome) followed a more relapsing and remitting course. Most patients had isolated stiffness and spasms of the lower limbs for several years, but, in time, about three quarters of the patients developed involvement of the upper limb, half developed mild sphincter disturbance (urinary frequency, urgency, and occasional urge incontinence) and almost 40% developed symptoms or signs of brainstem disturbance (often transient). The mean periods before involvement of the upper limbs, sphincters, or brainstem was 3, 5, and 2 years respectively. Patients with distal rigidity become more disabled over time, with six patients becoming wheelchair bound after a mean of 3.5 years, despite treatment. By contrast none of the patients with chronic axial stiffness (stiff man syndrome) became wheelchair bound. None of the patients with axial or distal stiffness developed any signs of an underlying malignancy.
INVESTIGATIVE FINDINGS
There were no significant differences in the routine haematological and biochemical blood tests, with the exception of the presence of diabetes mellitus in three patients with axial stiffness (stiff man syndrome; table 2). All but one of the patients with axial rigidity had antibodies to glutamic acid decarboxylase (GAD) in the serum, together with a range of other autoantibodies (including anti-islet cell, thyroid microsomal, gastric parietal cell, and smooth muscle antibodies). Most patients with distal stiffness (stiff limb syndrome), on the other hand, were anti-GAD antibody negative. The two patients in whom anti-GAD antibodies were present were otherwise indistinguishable from other patients with distal stiffness. Patients with distal rigidity also had a much lower incidence of other autoantibodies.
Investigative findings in stiff people
Just over half of the patients with axial rigidity (stiff man syndrome) had oligoclonal bands confined to CSF whereas only 17% of patients with distal rigidity (stiff limb syndrome) had such bands in their CSF. The index patient with progressive encephalomyelitis with rigidity had an abnormal CSF from the outset of his rigid syndrome, and as the disease progressed so did the abnormalities in the CSF with an increasing lymphocytosis and persistent oligoclonal bands with a raised protein concentration.
No abnormalities were found on MRI in the vast majority of patients. The brain abnormalities in the remaining patients consisted of one or a few non-specific white matter lesions (n=3), mesial temporal sclerosis in a young patient with epilepsy6 and global atrophy (n=1). The only abnormal spinal MRI was in a 37 year old woman with distal stiffness, in whom there were scattered white matter lesions within the cervical cord. The nature of these abnormalities was not clear, but her central motor conduction time and limb somatosensory evoked potentials (SEPs) were normal.
Routine peripheral neurophysiology showed no evidence of a neuropathy or neuromuscular problem; specifically, none of the patients had any evidence of neuromyotonia. The index patient with progressive encephalomyelitis with rigidity had profuse denervation in all four limbs. Central motor conduction time, SEPs from the lower limbs, and visually evoked responses were each abnormal in only one patient.
All patients with axial stiffness (stiff man syndrome) had continuous motor unit activity in their lumbar paraspinal muscles, with half having additional continuous motor unit activity in their proximal lower limbs. By contrast only four patients (31%) with distal stiffness (stiff limb syndrome) had continuous motor unit activity in their paraspinal muscles, whereas continuous motor unit activity was recorded distally in the affected leg or arm in all patients. Continuous motor unit activity tended to be segmented in half of the patients with distal stiffness. The cutaneomuscular reflexes from the tibial nerve at the ankle were virtually always abnormal in both groups with chronic stiffness. Similarly, about two thirds of patients in each group had reflex spasms to unexpected auditory stimuli. However, the nature of the EMG activity recorded during action and reflex induced spasms did serve to distinguish between the two groups (fig 1). In those patients with axial stiffness (stiff man syndrome) the EMG discharge was indistinguishable from a normal interference pattern, but in three quarters of patients with distal stiffness (stiff limb syndrome) the EMG was abnormally segmented, consisting of the grouped discharges of many motor units (often an exacerbation of the resting discharge pattern). Repetitive EMG bursts were irregular in all but three of these patients.
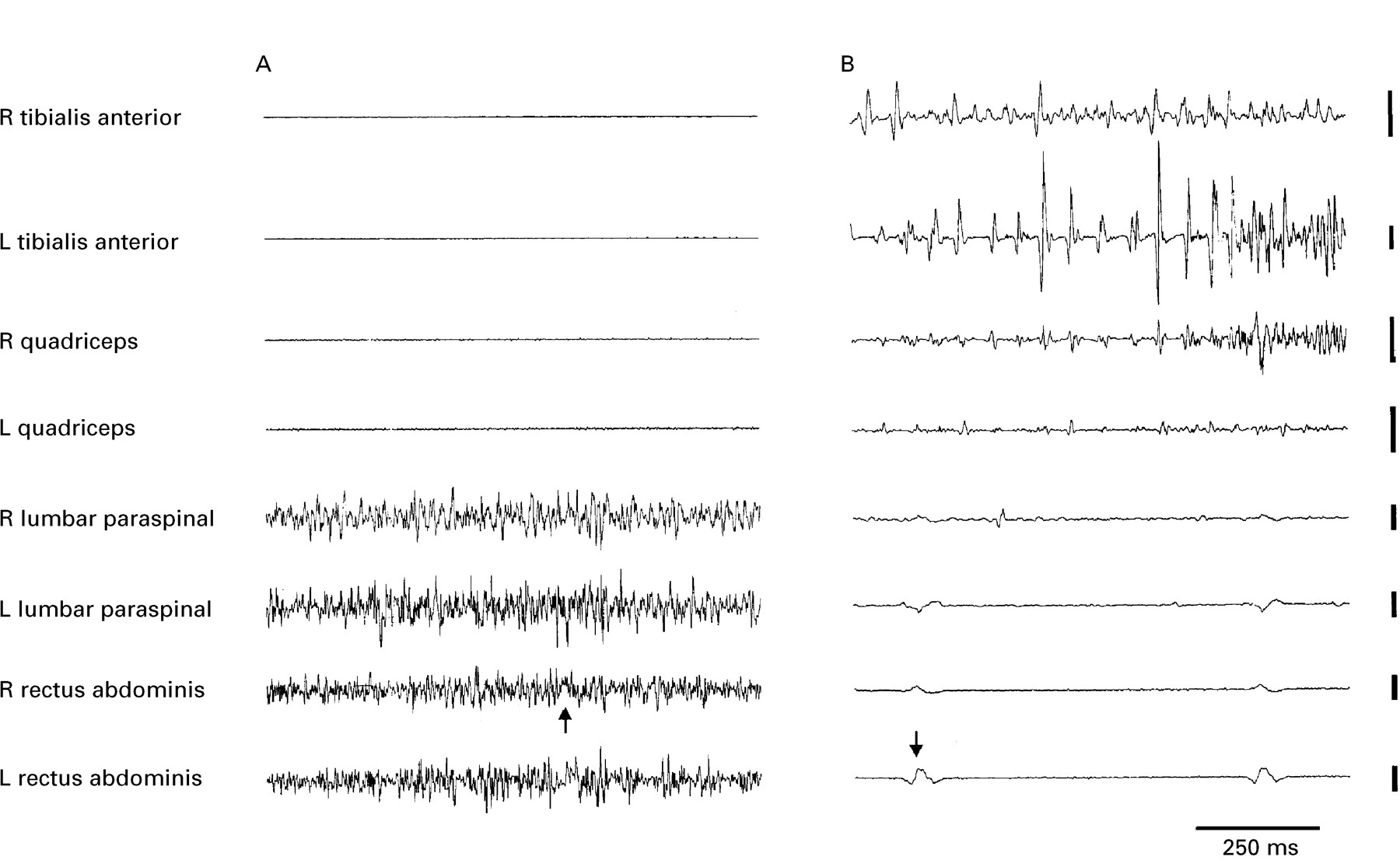
(A) Unrectified surface EMG activity during spontaneous spasms in a patient with chronic axial rigidity (stiff man syndrome) and positive anti-GAD antibodies, and (B) in a patient with rigidity of the distal lower limbs (stiff limb syndrome) who was anti-GAD antibody negative. (A) The spasm is confined to the muscles of the trunk, and EMG activity is indistinguishable from that recorded in a voluntary contraction. (B) The spasm is confined to the lower limbs, and EMG activity in the left (L) tibialis anterior tends to segment into large, but brief discharges. ECG artefact can just be made out in A and is more clearly visible in B (arrowed). Vertical calibrations are 100 μV and 500 μV for the lower and upper four channels respectively.
RESPONSE TO THERAPY
The index patient with progressive encephalomyelitis with rigidity failed to show any response to high dose baclofen and diazepam, as well as to high dose intravenous methylprednisolone, intravenous immunoglobulin, and plasma exchange. The other patient required diamorphine for control of his painful spasms.
Rigidity and spasms responded to a combination of baclofen and diazepam in all but one of the patients with axial stiffness (stiff man syndrome), the exception being the patient with negative anti-GAD antibodies. Mobility improved in half the patients, and treatment effects were sustained over time. By contrast, two patients with distal stiffness (stiff limb syndrome) did not respond to either or both drugs, and seven only had a partial response. Mobility only improved in a quarter of patients with distal stiffness.
The role of immunosuppression was less clear. Five patients with distal stiffness (stiff limb syndrome) were treated with either oral or intravenous steroids, and only one reported some slight benefit, with another patient describing a definite deterioration. One of the two patients with axial stiffness treated with steroids had a slight benefit, and the other reported no significant change in his condition. The two patients with axial stiffness (stiff man syndrome) treated with plasma exchange reported no benefit, whereas one patient treated with intravenous immunoglobulin responded dramatically.5 These treatments were not tried in those with distal stiffness (stiff limb syndrome).
PATHOLOGICAL FINDINGS IN A PATIENT WITH PROGRESSIVE ENCEPHALOMYELITIS WITH RIGIDITY
Only one patient came to postmortem. Both lungs were congested with patchy basal consolidation, confirming bronchopneumonia as the immediate cause of death. Thorough examination did not disclose an occult malignancy. The brain weighed 1490 g and had no atrophy or structural abnormality. Histologically there was chronic leptomeningitis, especially severe in the leptomeninges overlying the brainstem and spinal cord. There was also evidence for a florid subacute polioencephalitis with frequent cuffs of mature perivascular lymphocytes (mixed T and B cells), plasma cells, and macrophages, as well as neuronophagia, microglial nodules, and diffuse microglial activation accompanied by marked astrogliosis (fig 2). As a consequence there was variable neuronal loss in the affected areas. In the cerebral hemispheres the encephalitic process was most severe in the grey matter of the medial temporal lobe including the hippocampus and amygdala, the anterior cingulum, and insular cortex and similar, although less severe changes, were present in the grey matter of the thalamus, subthalamus, hypothalamus including the mamillary bodies, putamen, globus pallidus, and the nucleus basalis of Meynert. The brainstem showed extensive involvement: the periaqueductal grey matter, the third nerve nucleus, red nucleus, and substantia nigra were affected in the midbrain and the involvement of the periaqueductal grey matter, loci coerulei, nucleus centralis superior, nucleus centralis oralis, processus griseum pontis supralemniscalis, and the griseum pontis was apparent in the pons. The medulla was also among the anatomical areas which were most affected and both microglial nodules and cuffs of perivascular lymphocytes were seen to affect extensively the grey matter underlying the fourth ventricle. The structures involved included the hypoglossal nucleus, the dorsal motor nucleus of the 10th cranial nerve, the medial vestibular nucleus, and the lateral cuneate nucleus. In addition, the medullary reticular formation and inferior olive were also affected. In the lower medulla the hypoglossal nucleus was relatively mildly affected, as were the gracile and cuneate nuclei as well as the nucleus tractus spinalis trigemini. The cerebellum showed moderate Purkinje cell loss with basket cell preservation and gliosis, and microgliosis of the molecular layer. Microglial nodules were noted in the dentate nucleus of the cerebellum.

{kind=link}
{kind=link}
Histology of spinal cord and brain in a patient with progressive encephalomyelitis with rigidity. (A) Blood vessel in the hippocampus surrounded by thick lymphocytic cuff. (B) Microglial proliferation in the cervical cord. (C) Microglial proliferation in the amygdala. Haematoxylin and eosin preparation (A, B, and C). (D) Typical microglial module shown by Ham56 immunohistochemistry. Magnification originally×300 in A, B, and D, and×500 in C.
Several levels representing the cervical, thoracic, lumbar, and sacral cord were examined. All major areas of the spinal cord showed a considerable increase in number of micoglial cells in the grey matter with frequent formation of microglial nodules. The anterior horn cells showed, in general, mild depletion and the intermediolateral cell columns were involved at some levels. The neurons of Clarke’s column appeared preserved. Frequent, rather thick perivascular lymphocytic cuffs were present in the lateral and posterior columns at all levels and in the anterior column at some of the levels of the cord examined. The white matter was well myelinated at all levels examined. Special stains for bacteria and fungi were negative as were immunostains for herpes simplex virus, cytomegalovirus, and Toxoplasma gondii.
Autonomic ganglia (superior cervical ganglia, paravertebral sympathetic ganglia and celiac ganglion), showed variable infiltrates of chronic inflammatory cells within the ganglia in mainly perivascular and subcapsular locations. The infiltrates were composed of lymphocytes, plasma cells, and macrophages. This inflammatory process seemed particularly marked in the celiac ganglia where diffuse infiltrates of macrophages were present, surrounding ganglion cells with focal neuronophagia. Dorsal root ganglia at the cervical and thoracic levels disclosed a milder but similar inflammatory picture. It was difficult to assess the extent of nerve cell loss within these peripheral ganglia, but nodules of Nageotte were noted in increased numbers indicating focal depletion. However, the dorsal roots showed no significant depletion of myelinated fibres. Median nerve showed focal lymphocytic infiltrates in the endoneurium without any evidence of degeneration.
Discussion
We were able to identify 23 patients with rigidity and spasms in association with continuous motor unit activity at rest, in whom there was no evidence of neuromyotonia or extrapyramidal or pyramidal dysfunction. Only two of these patients had a subacute course, and postmortem confirmed progressive encephalomyelitis with rigidity in one. The remaining 21 chronic patients were divided into those whose major complaint was axial stiffness and spasm, or stiffness and spasm of a distal limb. It turns out that this simple division serves to identify two groups that can be distinguished in terms of clinical features, investigative findings, and response to therapy. Those with axial stiffness and spasms fulfilled the general criteria of Lorishet al for the diagnosis of the classic stiff man syndrome.7 The remainder had a condition akin to the stiff leg syndrome reported by Brown et al,4although here we show that this condition may also involve the upper limb and brainstem in some patients. Accordingly, we rename this the stiff limb syndrome.
PROGRESSIVE ENCEPHALOMYELITIS WITH RIGIDITY
Progressive encephalomyelitis with rigidity is a rare condition characterised by progressive rigidity of both limb and truncal muscles with brainstem involvement, myoclonus, and spasms.8-12Most patients have died within 2–3 years. Both our patients developed bulbar difficulties before the rapid and disseminated involvement of all four limbs and the trunk. One patient came to postmortem. The principal neuropathological findings were those of a subacute encephalomyelitis which primarily, but not exclusively, affected grey matter (polioencephalitis). Of the hemispheric structures the limbic areas were the most affected and the pathological process was also severe in the lower brainstem and spinal cord. In addition, there was ganglionitis affecting both the sympathetic and dorsal root ganglia. Although the anatomical distribution of the histological changes was consistent with a paraneoplastic encephalomyelitis,13 the presence of a primary malignancy could not be confirmed at postmortem—a situation not uncommonly found in more well recognised paraneoplastic syndromes.14
In our patient the histological changes were, in general, more widespread than in the other pathologically documented patients previously reported with progressive encephalomyelitis with rigidity.8-12 However, it is a common finding in all patients including ours that the lower brainstem and spinal cord is severely affected. Although the involvement of the anterior horn cells is documented in some of the patients, the depletion of the spinal inhibitory interneurons, specifically examined in one previous study, may be essential in the pathogenesis.12
CHRONIC AXIAL STIFFNESS AND SPASMS
These patients all had the classic stiff man syndrome as first described by Moersch and Woltman.15 This presents with paraspinal and abdominal rigidity with a lumbar hyperlordosis and superimposed spasms precipitated by sudden movement, noise, tactile stimulation, or emotional upset. It is associated with normal muscle strength and a normal sensory examination, but continuous motor unit activity is found in the paraspinal muscles. The proximal legs can be involved in the process, and this is often only apparent on walking when the patient has a stiff gait. The calf and foot muscles are rarely, if ever involved.7 16 There is no upper limb, cranial nerve, or sphincter disturbance.
All eight of our patients showed these classic features of stiff man syndrome, with the appropriate neurophysiological abnormalities of continuous motor unit activity and abnormal exteroceptive reflexes. All but one of our patients had anti-GAD antibodies, a figure that is higher than that seen in other studies,2 3 perhaps because of our insistence that rigidity and spasms should predominantly involve the trunk. The association with insulin dependent diabetes mellitus and other autoantibodies was confirmed, with 37.5% of the patients having insulin dependent diabetes mellitus and 87.5% having other autoantibodies including anti-islet cell antibodies.17 18 Most patients responded to a combination therapy of diazepam and baclofen, but the response to immunotherapy was disappointing, except in one patient. Our patients with stiff man syndrome had a typical natural history of slow progression without obvious spontaneous relapses or remissions. None became wheelchair dependent, in agreement with earlier studies.19
The pathophysiology of stiff man syndrome is unclear. No histopathological abnormalities have been identified in typical patients.20 An autoimmune basis is thought likely in view of the autoantibody profile of many of the patients.2However, it is unclear whether anti-GAD antibodies are central to the pathogenesis of this condition or an epiphenomenon.
CHRONIC STIFFNESS AND SPASMS OF THE DISTAL LIMB
Thirteen patients had stiffness and spasms dominating in one or more distal limbs, as part of an illness lasting up to 19 years. Truncal rigidity and/or hyperlordosis of the lumbar spine were only present in two of these patients, and in these instances were overshadowed by rigidity elsewhere.
Four of the patients have been previously reported in detail by Brownet al, who described a chronic progressive disorder beginning in middle age and involving stiffness and painful spasms of the lower limbs.4 Symptoms and signs of brainstem, pyramidal, and sensory dysfunction were absent, and sphincter disturbance developed after many years in only one patient. Likewise, there was little or no electrophysiological evidence of long tract disturbance. However,continuous motor unit activity was present at rest in at least one limb muscle, cutaneomuscular reflexes were abnormal, and spasms had a characteristic segmented appearance on EMG. Brownet al suggested that the disorder was due to a chronic spinal interneuronitis. Here we extend the range of this disease, which may begin in the upper limb or lead to late brainstem dysfunction in the minority of patients.
Patients typically presented between the ages of 20 and 50 with a rigid distal limb, usually the lower leg. About half of the patients followed a relapsing and remitting course, and around a third developed symptoms or signs of brainstem involvement. These were usually delayed in onset and transient. Similarly, half developed a sphincter disturbance after a mean of 5 years. None of the patients developed the generalised myoclonus characteristic of the jerking stiff man syndrome.21 Cortical and cognitive deficits were absent, as were seizures, with one exception. Half the patients become wheelchair or bed bound after a few years, in marked contrast with those with stiff man syndrome.
The autoimmune profile of patients with stiff limb syndrome also differed from those with stiff man syndrome. There were no patients with insulin dependent diabetes mellitus, and serum autoantibodies were present in only around a third of patients. In addition, only 15% had anti-GAD antibodies, as opposed to 88% of our patients with stiff man syndrome. Patients with stiff limbs also had a lower incidence of intrathecal synthesis of immunoglobulin (17% v 57% of patients with stiff man syndrome), although the total CSF protein was more often raised.
Detailed neurophysiological studies in these patients support the contention that patients with stiff limbs are different from those with stiff man syndrome. Although both groups had continuous motor unit activity at rest, the distribution was very different. In addition, three quarters of patients with stiff limbs had spasms with a characteristic interference pattern, consisting of hypersynchronous segmented discharges. This was not seen in the stiff man syndrome.
These patients therefore have a distinctive clinical and electrophysiological picture. Despite the severity of the limb stiffness and spasms there was a striking scarcity of specific symptoms, signs, or electrophysiological abnormalities attributable to the long tracts of the spinal cord. This has led to the suggestion that the rigidity and the spasms evoked by peripheral somaesthetic stimuli, voluntary action, or startle are due to a localised spinal interneuronitis.4 To date, histology has been reported in only one patient with a clinical history consistent with the stiff limb syndrome, and showed an encephalomyelitis predominantly affecting the lumbar cord.22 Indeed patients with the idiopathic stiff limb syndrome are very similar to other patients with known focal pathology preferentially involving the grey matter of the spinal cord. Thus intrinsic tumours,23 syringomyelia,24vascular insufficiency,25 and paraneoplastic myelitis26 may be associated with continuous motor unit activity, and reflex and action induced spasms of the trunk and lower limbs. The relatively selective destruction of spinal interneurons in some of these patients has been confirmed histologically.12 23 Effective stimuli need not be restricted to somaesthetic stimulation below the level of the spinal cord pathology. Paradoxically, jerks and spasms of the legs can be precipitated in patients with spinal lesions by startle inducing stimuli, including sounds.25 27 Presumably this represents an excessive response at the segmental level to descending reticulospinal activity.
Although, with one exception,22 pathological reports are lacking in the stiff limb syndrome, the condition has many clinical similarities with the jerking stiff man syndrome, in which there is increasing evidence for a polioencephalomyelitis largely indistinguishable from that found in progressive encephalomyelitis with rigidity.3 28 Patients with the jerking stiff man syndrome can survive more than 10 years,21 and may have evidence of an autoimmune diathesis.29 Rigidity and spasms often start in and sometimes remain confined to the limbs in this condition.28 However, unlike the stiff limb syndrome, the clinical picture in these patients is dominated by marked cranial nerve signs and a characteristic brainstem myoclonus that involves all four limbs.21 Perhaps, the essential difference between these patients and the stiff limb syndrome lies in the distribution rather than the nature of the pathology.
Occasionally, rigidity of the limbs (sometimes with later spread to the trunk) is seen in the setting of breast or small cell lung carcinoma. Such patients have antibodies against the vesicle associated protein amphiphysin.30-32 Many of the patients in our series presented before the identification of this antibody,30and none of our patients were tested for it. Nethertheless, the long duration of the stiff limb syndrome in our series would be very much against a paraneoplastic aetiology.
Conclusion
Of our 23 patients about a third had stiff man syndrome, and two had an aggressive progressive encephalomyelitis with rigidity. However, most had a distinct clinical entity that we term the stiff limb syndrome. Such patients present with rigidity and painful spasms of a limb, usually the distal leg, and abnormal fixed posturing of the foot or hand. There is sparing of the truncal musculature, at least in the early stages of the disease, which may follow a relapsing and remitting course. There may be involvement of the brainstem and sphincters, but this is usually delayed by several years. Neurophysiologically, such patients have continuous motor unit activity in the affected limb with abnormal exteroceptive reflexes, and a characteristic interference pattern during spasms. Most patients have neither anti-GAD nor other autoantibodies. The response to diazepam and baclofen is only partial, with half of the patients becoming wheelchair bound. The role of immunosuppressive therapy is currently unknown. The disease is indolent, lasting many years, and may be due to a spinal interneuronitis, although this remains speculative.
Acknowledgments
We thank the following for referring some of the patients: Dr L Findley, Dr RS Kocen, Professor CJ Mathias, Professor WI McDonald, Dr JA Morgan-Hughes, Dr P Rudge, Professor M Ron, and Dr J Duncan. We also thank Dr J Land and Dr M Hawa for performing the serological testing, and Dr PD Thompson and Dr JC Rothwell for their help with some of the electrophysiological recordings.