Article Text

Abstract
We present a family in which an initial clinical diagnosis of autosomal dominant pure hereditary spastic paraparesis (HSP) was made on the basis of a three generation pedigree in which both males and females presented with a spastic paraparesis. Subsequent biochemical and genetic analysis revealed that the family was in fact affected by the adrenomyeloneuropathy subtype of X-linked adrenoleukodystrophy. In the family described, both males and females were affected by a spastic paraparesis, and there was no male to male transmission, consistent with both autosomal dominant and X-linked inheritance. This report illustrates the importance of assaying very long chain fatty acids (VLCFAs) in any HSP family where there is no male to male transmission.
- X-linked adrenoleukodystrophy
- hereditary spastic paraparesis
- ALD, adrenoleukodystrophy
- AMN, adrenomyeloneuropathy
- HSP, hereditary spastic paraplegia
- MRI, magnetic resonance imaging
- VLCFAs, very long chain fatty acids
Statistics from Altmetric.com
- ALD, adrenoleukodystrophy
- AMN, adrenomyeloneuropathy
- HSP, hereditary spastic paraplegia
- MRI, magnetic resonance imaging
- VLCFAs, very long chain fatty acids
INTRODUCTION
The hereditary spastic paraplegias (HSPs) are a group of neurodegenerative conditions that share the principal clinical feature of progressive lower limb spastic paralysis caused by either failure of development or progressive degeneration of the corticospinal tracts. They are subdivided into pure and complicated forms, depending on the presence of additional neurological or non-neurological features.1,2
Pure HSP is the largest HSP subgroup and most of these families show autosomal dominant inheritance, with autosomal recessive inheritance accounting for nearly all of the remainder. There are eight known loci for autosomal dominant pure HSP, and four of the causative genes have been identified. Mutations in two of these genes, spastin and atlastin, account for around 50% of autosomal dominant pure HSP families.1,2 However, in families lacking a mutation in one of these genes, diagnosis relies on clinical criteria,2 which emphasise the fact that pure HSP is a diagnosis of exclusion, and that other potential diagnoses should be sought before arriving at this diagnosis. We present a family in which an initial clinical diagnosis of autosomal dominant HSP was made on the basis of a three generation pedigree in which both males and females presented with a spastic paraparesis. However, subsequent biochemical and genetic analysis confirmed that the family was in fact affected by X-linked adrenoleukodystrophy (ALD). This case therefore illustrates the critical importance of testing for X-linked ALD in every family with apparent pure HSP in which there is no male to male transmission.
CLINICAL HISTORY
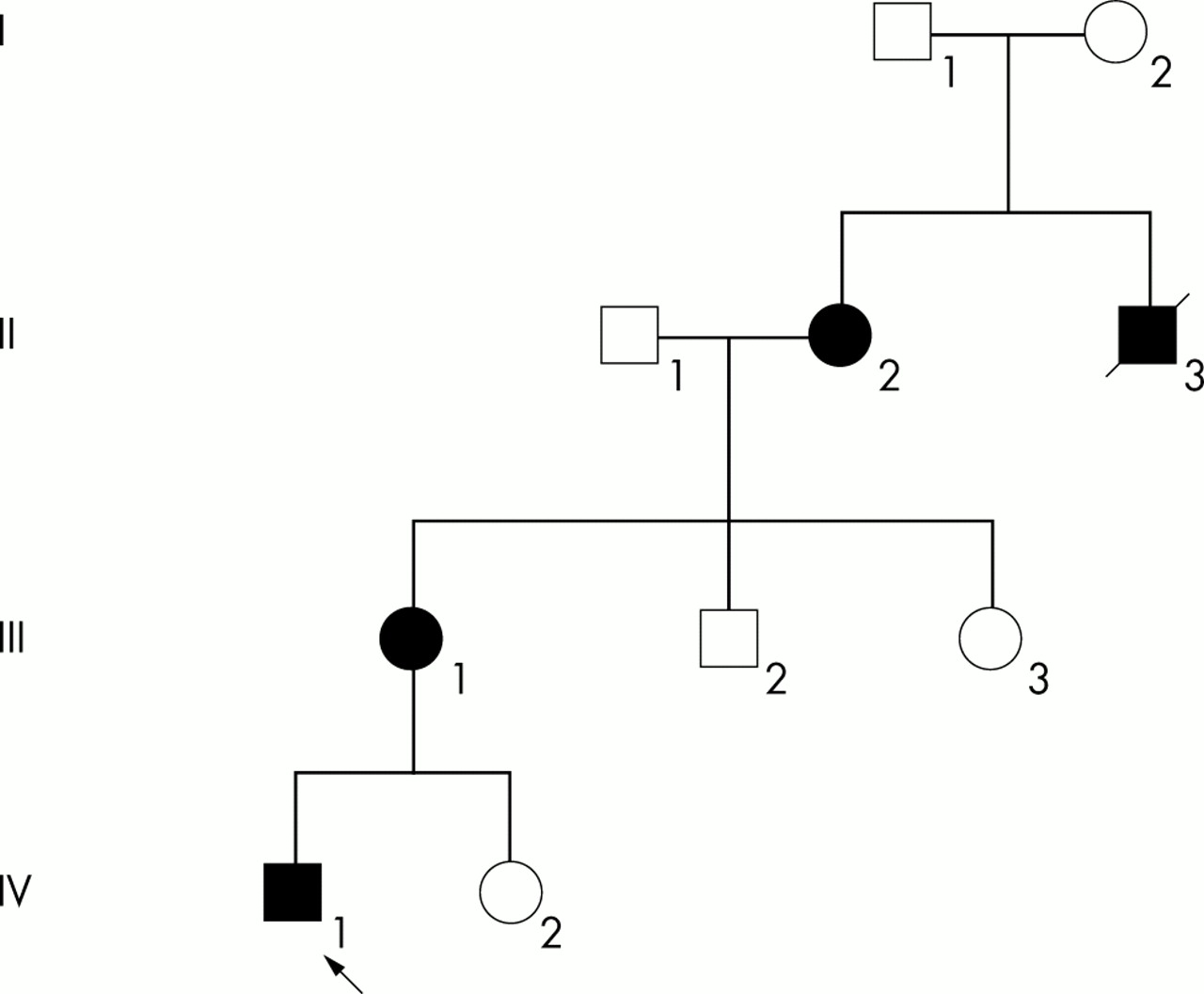
The family relationships are shown in the pedigree (fig 1).

{kind=link}
Family tree. Please see text for the description of each case. The consultand is indicated by an arrow. Filled symbols indicate both molecularly confirmed and probable X-linked adrenoleukodystrophy.
Case 1 (IV1)
A 26 year old man presented to the neurology service with an 18 month history of progressive difficulty in walking, and a more recent history of inability to run. There were no sensory and urinary symptoms. Neurological examination of the cranial nerves and upper limbs was normal, whereas in the lower limbs, tone was markedly increased, although full power was retained. Lower limb reflexes were very brisk with sustained clonus and upgoing plantar responses bilaterally. Sensation was normal in all modalities.
Initial investigations including full blood count, erythrocyte sedimentation rate, electrolytes, glucose, liver function tests, thyroid stimulating hormone (TSH) immunoglobulins, autoantibody screen, chest x ray and electrocardiogram were all reported as normal. Specific neuroimaging with magnetic resonance imaging (MRI) of the brain and cervical and thoracic spinal cord revealed no pathology. Cerebrospinal fluid protein was mildly elevated at 0.54 g/dl; no oligoclonal bands were present.
Case 2 (III1)
A 54 year old woman presented to the neurology service with her son (patient IV1). She gave a history of clumsiness and recurrent falls. There were no bladder or sensory symptoms. Physical examination revealed a spastic paraparesis with some mild asymmetry, being more marked on the left side. Examination of cranial nerves and upper limbs was normal.
Case 3 (II2)
This was an 82 year old woman, the mother of III1 and grandmother of IV1. She reported longstanding difficulties with walking but had not formally sought medical advice.
Case 4 (II3)
This man, the brother of case II2, died in 1979 of renal failure. He had always had some gait difficulty, walking on tiptoe with high foot arches. He is presumed to have been affected by the same neurological condition, although definitive evidence for this is lacking.
In light of the clinical features and apparent transmission pattern in the family, a presumptive diagnosis of autosomal dominant pure HSP was made and the proband was referred to the clinical genetics service for counselling on this basis. However, in parallel with this a venous blood sample was sent for analysis of very long chain fatty acids (VLCFAs) to investigate the possibility of an alternative neurological diagnosis.
The results of the VLCFA analysis strongly suggested a diagnosis of ALD (normal ranges in parentheses): decosanoate (C22): 41 μmol/l (30–112); tetracosanoate (C24): 58 μmol/l (14–80); hexacosanoate (C26): 2.5 μmol/l (0.33–1.5); ratio C24/C22 1.41 (0.44–0.97); ratio C26/C22 0.061 (0.005–0.030). In view of this new information, genomic DNA from the proband was screened for mutations in the ALD gene. A missense mutation 2262G>A, previously reported to be pathogenic based on functional studies,3 was identified in exon 9 of the ALD gene, confirming X-linked ALD as the correct diagnosis in this family. Further testing revealed that both the patient’s mother and maternal grandmother carried the same mutation. The proband (IV1), his mother, and his maternal grandmother were referred for assessment of adrenal function. Normal results were obtained for each individual.
DISCUSSION
We present a family in which X-linked ALD presented with features that led to an initial diagnosis of autosomal dominant HSP. X-linked ALD (OMIM 300371) is a disorder of the nervous system white matter and adrenal cortex, with an estimated incidence of approximately 1 in 50 000.4 There are four main clinical subgroups in affected males. The childhood cerebral form manifests usually between the ages of four and eight years with progressive impairment of cognition, hearing, vision, and motor function. It is the most severe phenotype. Adolescent ALD is similar to the childhood form, but onset is typically between the ages of 10 and 21 years. The third phenotype, adrenomyeloneuropathy (AMN), manifests most commonly in the late twenties as progressive paraparesis, sphincter disturbance, and distal sensory loss. It is slowly progressive over decades. The fourth phenotype presents with primary adrenal insufficiency at any age after two years, with no evidence of neurologic abnormality. These clinical subgroups do not breed true within families, and different phenotypes are often found within a single family.
Although ALD is inherited in an X-linked pattern, carrier females do exhibit symptoms and signs of the condition, typically of the AMN phenotype and sometimes these features can be accompanied by adrenal failure.5 In one series,6 61% of carrier females had some neurological abnormality resembling AMN, with severe disability in 14%. The mean age at onset of symptoms was 37.8 years, later than the typically third decade onset found in males with the AMN phenotype. Therefore, as far as the AMN phenotype is concerned, ALD can be considered to be an X-linked dominant condition with reduced penetrance and a milder phenotype in females. This does mean that it is possible for X-linked pedigrees to resemble closely autosomal dominant pedigrees, as in this case. Other instances have been previously reported.7
The possibility for confusion is compounded by the fact that a distinction between AMN and uncomplicated HSP can not be made on clinical grounds. In both, a spastic paraparesis occurs and may be associated with urinary symptoms and mild sensory symptoms and signs in the lower limbs. In HSP, MRI of the brain and spinal cord are usually normal, though atrophy of the thoracic spinal cord has been observed in some subjects.8 Similarly, MRI abnormalities of the brain are often absent in AMN. Brain MRI was normal in 56% of men and 80% of women in a series of 119 men and 45 women with the AMN phenotype.9 Brain parenchymal abnormalities resembling those seen in childhood-onset ALD are reported in the remainder.
The plasma concentration of VLCFAs is elevated in more than 99% of males with X-ALD, regardless of symptoms; this assay has a sensitivity of approximately 85% in female carriers.10 Mutation analysis of the ABCD1 gene, mutated in ALD, has a sensitivity of well over 90%.11
Neurophysiological studies should also be interpreted with caution. Apparently “pure” HSP families may have abnormal nerve conduction velocities.12 In the AMN subtype of ALD, while abnormalities of peripheral nerve function are frequent, they are not a universal finding.13
In the present case, the correct diagnosis of X-linked ALD was made because the appropriate diagnostic test was done, but it is clear that X-linked ALD continues to be underdiagnosed.3 This is particularly relevant as the severe childhood form of the condition can result from the same mutation that underlies AMN, and the consequences for genetic counselling of incorrect diagnosis are therefore very significant, even tragic. Therefore, X-linked ALD should be considered in the differential diagnosis of any HSP, where male to male transmission is absent, and in apparently sporadic cases of spastic paraparesis.
REFERENCES
Footnotes
-
Competing interests: none declared