Article Text

Abstract
Objective: To compare the efficacy and safety of clozapine in drug induced psychosis in Parkinson’s disease (PD).
Methods: A four week, randomised, double blind, parallel comparison of clozapine and placebo, followed by a 12 week clozapine open period, plus a one month period after drug discontinuation, in 60 patients with PD. The primary efficacy outcome was the “clinical global impression scale” (CGI); the positive subscore of the “positive and negative syndrome scale” (PANSS) was used as the secondary efficacy parameter and the “unified Parkinson’s disease rating scale” (UPDRS) and the “mini mental test examination” (MMSE) as safety outcomes.
Results: The mean (SD) dosage of clozapine was 35.8 (12.5–50) mg at the end of the double blind period. The mean (SD) scores on the CGI improved by 1.8 (1.5) for the clozapine group compared with 0.6 (1.1) for the placebo group (p = 0.001). The mean (SD) positive subscore of PANSS improved by 5.6 (3.9) for the clozapine group (0.8 (2.8) for the placebo group; p < 0.0001). At the end of the open period, 25 patients had completely recovered from delusions and hallucinations, and 19 experienced a relapse within one month after the clozapine washout period. The UPDRS motor and MMSE mean scores did not change significantly in either group. Somnolence was more frequent with clozapine than with placebo.
Conclusions: Clozapine at a mean dose lower than 50 mg/day improves drug induced psychosis in PD without significant worsening of motor function, and the effect wears off once the treatment stops.
- Parkinson’s disease
- psychosis
- clozapine
- CGI, clinical global impression scale
- MMSE, mini mental test examination
- PANSS, positive and negative syndrome scale
- PD, Parkinson’s disease
- UPDRS, unified Parkinson’s disease rating scale
Statistics from Altmetric.com
- CGI, clinical global impression scale
- MMSE, mini mental test examination
- PANSS, positive and negative syndrome scale
- PD, Parkinson’s disease
- UPDRS, unified Parkinson’s disease rating scale
The occurrence of psychotic symptoms (disturbance of perception and thought) induced by dopaminergic drugs marks a new phase in the course of Parkinson’s disease (PD).1,2 To know whether psychosis is induced by drugs or not is difficult because of confounding factors. However, the term drug induced psychosis may be used when other significant psychiatric diseases are excluded in patients with no history of psychosis. Moreover, because all patients with PD received dopaminergic drugs, we have no means of distinguishing between the drugs or the disease as the prevalent aetiology. Not only do psychotic symptoms have a major impact on patients and their caregivers in dramatically limiting treatment possibilities, but they are also a risk factor for nursing home transfer and subsequent increased mortality.3 The prevalence of dopaminomimetic psychosis varies from 5% to 20%, depending on the methodology of the study and nature of symptoms taken into account.2 A recent community based study found that 16% of patients had hallucinations and delusions, and that age, late disease stage, institutionalisation, cognitive decline, and depression were significant risk factors.4 Several approaches have been proposed to manage these symptoms, including attempts to withdraw dopaminergic agonists and gradually decrease the levodopa drug “dose” and the use of a “low potency” neuroleptic treatment. However, the neurologist often faces a “motion emotion” dilemma,2 because these approaches can lead to unacceptable motor deterioration.5,6 Since the early study of Scholz and Dichgans,7 at least 32 open labelled trials involving more than 300 patients have suggested the usefulness of the atypical neuroleptic clozapine as a unique possibility of controlling drug induced psychosis, without compromising motor function, in many patients with PD.8 The efficacy and tolerability of low dose clozapine in the treatment of drug induced psychosis in PD has recently been outlined in the first double blind, placebo controlled study.9 Here, we report in detail the results of a large, multicentre study, including three consecutive, double blind, open and washout periods of clozapine treatment. The first double blind, placebo controlled period has been briefly reported.10
METHODS
This prospective, double blind, placebo controlled study was conducted at 13 centres in France between January 1996 and October 1997, in compliance with good clinical research practice guidelines and the Helsinki Declaration. The protocol was approved by the ethics committee of Grenoble and all patients gave their written consent.
Patient selection
Sixty patients suffering from idiopathic PD according to the “UK Parkinson’s Disease Society brain bank clinical diagnostic” criteria and experiencing a drug induced psychosis of at least two weeks’ duration were eligible for recruitment. All patients had failed to respond to standard therapeutic management; that is, no improvement of psychiatric symptomatology or an unacceptable worsening in motor symptoms despite the interruption of all anticholinergic agents (including tricyclic antidepressants), in addition to amantadine and selegiline, an attempt to withdraw dopaminergic agonists, and a progressive reduction in the doses of levodopa.
The psychotic symptom score had to be ⩾ 4 for at least one of the items P1 (hallucinations) or P3 (delusions) of the positive subscore of the “positive and negative syndrome scale” (PANSS).11 In addition, patients had to score > 3 on the “clinical global impression scale” (CGI).12 Both of these scales rate the severity of psychotic symptoms from 0 (no symptoms) to 7 (the most severe symptoms). Furthermore, at least 20 had to be scored on Folstein’s “mini-mental state evaluation” (MMSE).13 A history of recent changes was obtained from the patients. Patients were excluded if they had a history of medical conditions or drug treatment that might put them at special risk or bias the assessment of their clinical or mental status. Patients likely to require continuous treatment with drugs that can lower the white blood cell count, and those previously treated with clozapine, were also excluded, as were women of childbearing potential who were not practising a medically approved form of birth control.
Overall design and outcome measures
Four periods were planned in our study. The first (period I) was a period of screening. After a minimum seven day period of stability with levodopa and dopaminergic agonists at the minimal tolerable dose, eligible patients were randomised to receive clozapine or placebo once daily at bedtime (1/1 randomisation, block size of four). The second period (period II) of four weeks (day 0 to day 28) involved clozapine dose titration according to the following schedule: a starting daily dose of 6.25 mg, followed, if necessary, by progressive dose increases (maximum of three 12.5 mg steps each week) up to a maximum daily dose of 50 mg, which could not be reached within less than 10 days. The doses of antiparkinsonian drugs remained unchanged. The dose of clozapine could be reduced if adverse effects occurred by steps of 12.5 mg. The speed of changes was adapted to adverse effects. Antidepressant (tricyclics excepted) and benzodiazepine cotreatments were permitted both at baseline and during the trial, as long as they were used at the lowest possible dose and kept constant throughout the study. All patients who completed period II and those experiencing no improvement in items P1 and P3 of PANSS after two weeks of treatment (day 14) entered a 12 week unblinded open label period (period III), where they received clozapine. Because the randomisation code was not broken, the patients had the same dose escalation as during period II. However, from the third week of period III, the clozapine daily dose could be further increased up to a maximum of 100 mg/day. One week after the end of clozapine dose adjustment, an increase in the dose of antiparkinsonian drugs was also permitted if the patient’s mental status was normalised on the basis of items P1 and P3 of PANSS. However, simultaneous alterations in antiparkinsonian medications and clozapine dosages were forbidden. At the end of period III, patients demonstrating mental normalisation (PANSS items P1 and P3, < 3) were subjected to clozapine withdrawal within one week, and to a further three week follow up (period IV). The patients who could not be exposed to this withdrawal or those who relapsed within period IV were included in an extension protocol.
Treatment effects on psychiatric symptoms were assessed on a weekly basis in period II, every two weeks during the first month of period III, and then monthly, using the CGI, which was chosen as the primary efficacy parameter, and the positive subscore of PANSS (in which scores range from 7 to 49). Motor functions were assessed during “on” periods obtained by asking the patients to take their regular dose of levodopa before the consultation, which was scheduled at the same hour of the day and the same latency from levodopa intake throughout the study duration. We used the “unified Parkinson’s disease rating scale” (UPDRS) total score (part I + II + III; scores range, 0–166) and motor score examination (part III; scores range, 0–108) at baseline, at the end of period II and period III, and in cases of premature discontinuation. Because of the variability of UPDRS evaluations during “on” periods, the Schwab and England’s activities of daily living scale (UPDRS part VI, with a range of scores from 0 to 100, lower scores indicating more severe disability), assessed weekly, was chosen as the criterion to detect a possible worsening of parkinsonism: a 20% decrease in the UPDRS part VI up to day 14, or a 10% decrease thereafter entailed the interruption of clozapine dose escalation. Blood samples were taken weekly and physical signs were recorded daily for the first four days in periods II and III, and then on a weekly basis.
Randomisation and blinding
Randomisation was performed by the biostatistics department of Sandoz (subsequently Novartis), France using a validated system that automates the random assignment of treatment groups to randomisation numbers. Period II was double blind. The blinding was maintained when patients entered period III and received clozapine open labelled. Blood count results were not blinded because of the very low number of patients expected to suffer from agranulocytosis.14
Sample size determination
Because of the absence of bibliographical data at the time of study planning, sample size determination was empirical, based on practical considerations (feasibility) and previous experience. Sample size was set to 60 evaluable patients (that is, randomised patients completing period II).
Statistical analysis
Data were analysed separately for periods II, III, and IV. In period II, treatment groups were defined by randomisation and formal statistical testing was carried out. Safety analysis was based on all randomised patients having received at least one dose of study drug. Efficacy analysis was based on the “intention to treat” population, defined as all randomised patients having received at least one dose of study drug, with at least one assessment of PANSS positive items after drug intake. Major time points were defined as baseline and end of period II (last observation in period II). For efficacy parameters (CGI and PANSS) and some safety data (MMSE, UPDRS total and subscores), treatment groups were compared for changes from baseline to end of period II by means of the t test. Period III data (CGI, PANSS, MMSE, and UPDRS) were broken down into the two period II treatment groups and were also presented globally as a whole. Only the number of relapses was presented in period IV. Period III and IV data were mainly descriptive: within group comparisons were carried out for efficacy parameters. Other safety data (adverse events, abnormalities in white blood cell/neutrophil counts, and hypotension) were described in terms of frequency tables of treatment emergent events. With regard to adverse events codification, it was decided to pool any possible related symptom (akinesia, motor blockade, freezing, motor worsening, and decrease in length of phases “on”) under the preferred term “worsening of parkinsonism”.
Safety committee
A safety monitoring committee, composed of three physicians who did not otherwise participate in the study, met regularly. The committee reviewed all serious adverse events reports and its members had access to treatment codes.
RESULTS
Patients
In total, 60 patients were randomised. Figure 1 presents the numbers of patients in each study group and in each period II and III with reasons given for study discontinuation.
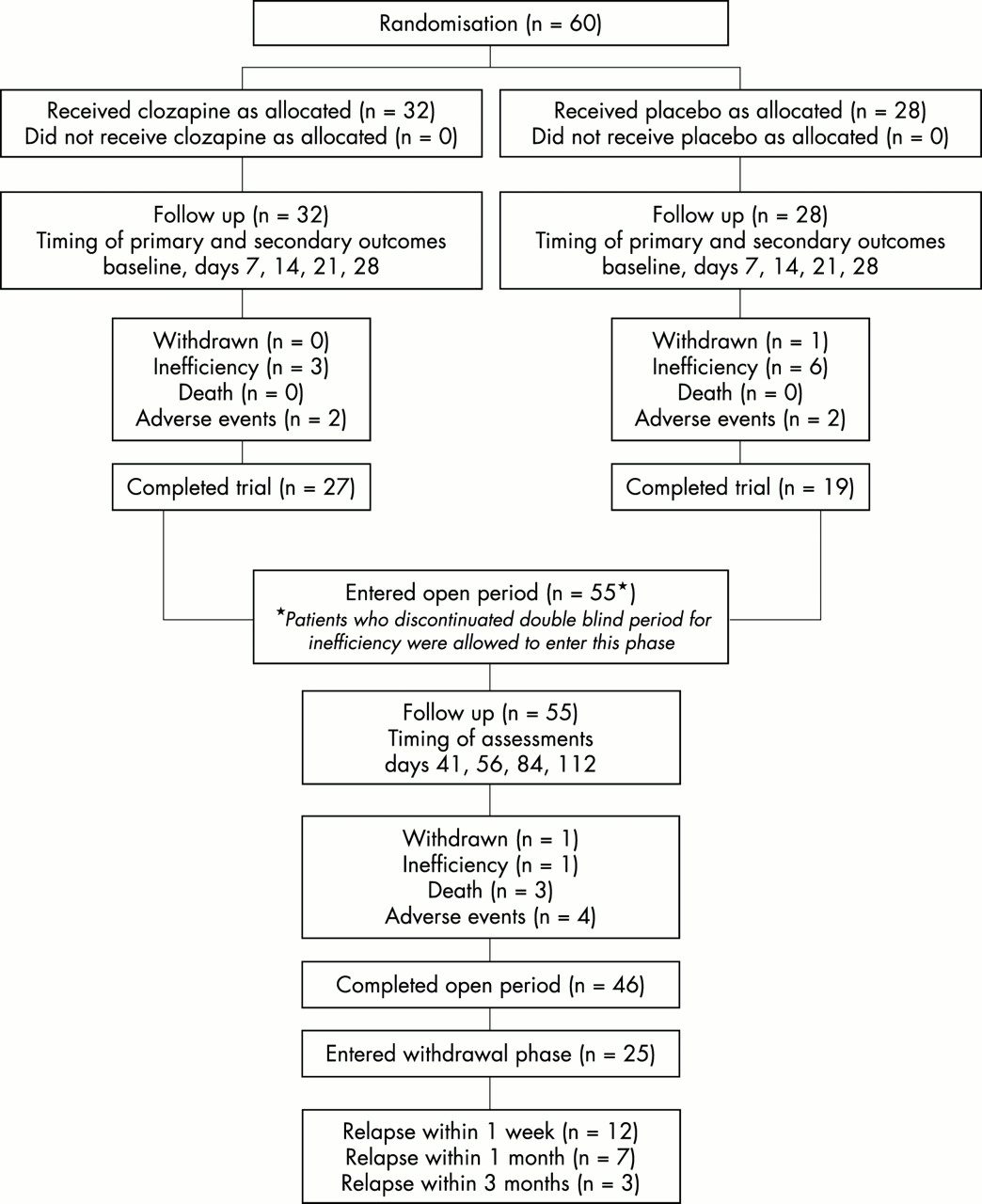
Flow diagram for randomised comparison of clozapine (left) versus placebo (right).
The double blind period was completed by 46 of the patients. Because all discontinuations as a result of inefficacy in period II occurred after the end of week 2, the patients could be entered directly into period III as planned by the protocol. Thus, 55 of the 60 patients were included in this open period, which was completed by 46 of them.
Table 1 presents the characteristics of the population and their baseline psychiatric assessments. There were no noticeable differences between the clozapine and placebo group with regard to disease history or severity, apart from a significantly higher MMSE score in the clozapine group (26.1 v 24.1; p = 0.01). With one exception, all patients were receiving levodopa and 14 were taking a dopaminergic agonist. Analysis of the baseline values of the PANSS positive score items shows that hallucinations (P3) and delusions (P1) were the major symptoms, whereas “grandiosity” was absent (mean score, 1).
Patient characteristics at baseline
Dosages
By the end of period II, patients were receiving a mean dose of 35.8 (range, 12.5–50) mg/day of clozapine or 41.7 (range, 6–50) mg/day of placebo. By the end of period III, clozapine was given at a mean dose of 40.0 (range, 12.5–100) mg/day in the group previously receiving clozapine and 42.5 (range, 6.25–100) mg/day in the group previously receiving placebo.
Efficacy
Tables 2 and 3 show the changes in efficacy parameters (CGI, positive subscore of PANSS) compared with baseline.
Comparison of change scores in patients taking clozapine compared with the placebo group at the end of period II (PII)
Comparison of change scores in patients taking clozapine compared with the placebo group at the end of period III (PIII)
Period II
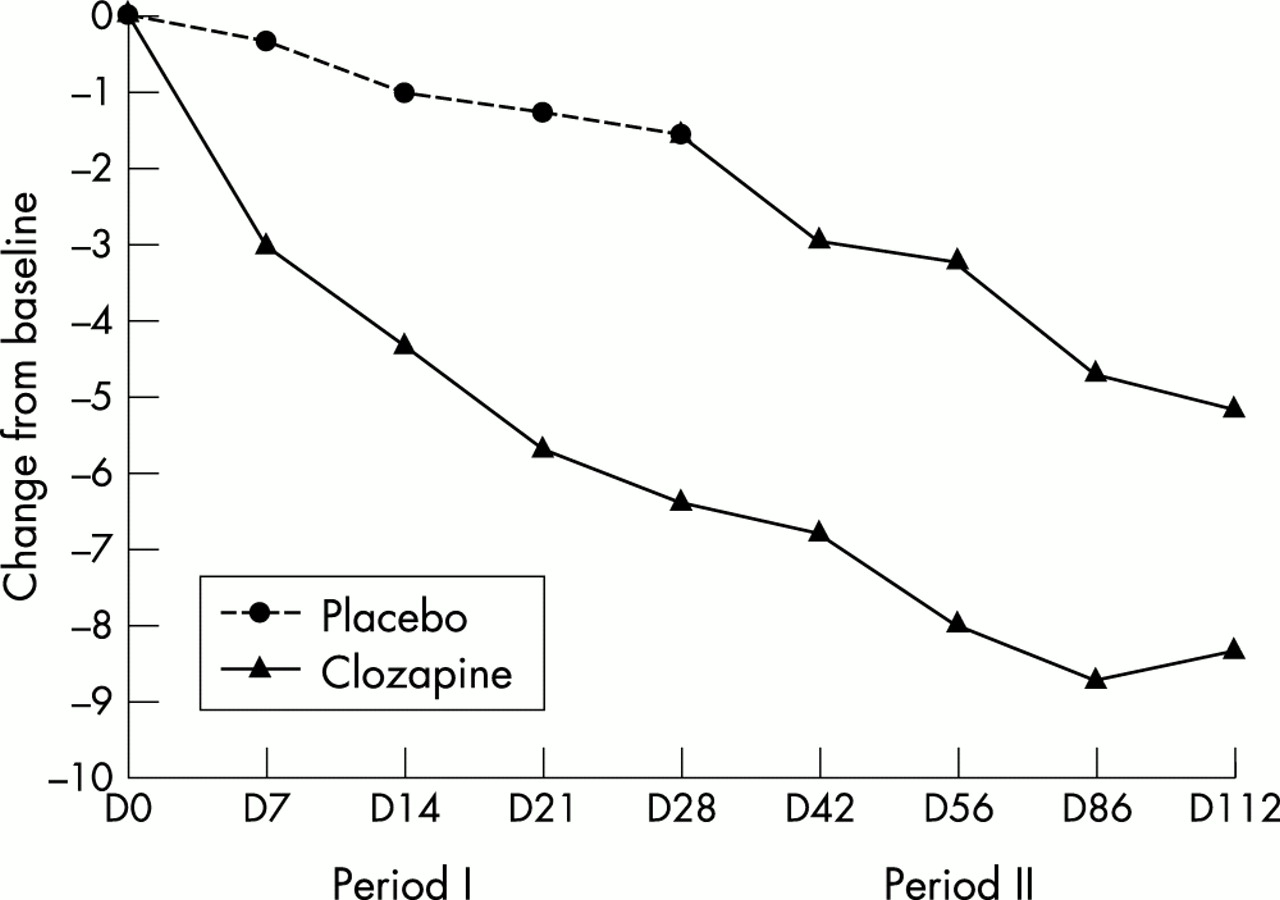
The improvement in the CGI and the PANSS global score was significantly greater in the clozapine group than in the placebo group. The effect was present as early as the first week of treatment (fig 2). All items of the positive subscore of PANSS, except grandiosity, were improved by clozapine when compared with the placebo (data not shown).
{kind=link}
{kind=link}
Time–efficacy relation: change from baseline (positive and negative syndrome scale). For the time–efficacy results, the analysis was performed on the patients who completed the period: observed cases analysis.
Period III
Those patients who had formerly received the placebo showed a similar improvement to those patients on clozapine during period II (table 3; fig 2). Those who had first received clozapine continued to improve, reaching a maximum between the second and the third treatment months.
At the end of period III, 25 patients (just over half of those completing this period) had recovered completely from delusions and hallucinations (items P1 and P3 of PANSS, < 3). Mean (SD) daily doses of levodopa were 737 (399) mg at the beginning of period III and 793 (407) mg at the end of this period, which represents an average increase of 56 (181) mg. This average change proved to be greater in those patients first allocated to the clozapine group (mean, 93; SD, 209 mg). This daily dose of levodopa had to be increased in 17 patients (by at least 250 mg in seven cases), but was reduced in five patients. In three patients a dopaminergic agonist was introduced or its dose increased.
Period IV
Drug withdrawal was attempted in 25 patients who had completely recovered from psychosis at the end of period III. A relapse was observed in 19 of these patients during period IV, as early as the first week in 12 patients. A subsequent follow up showed that three more patients relapsed within the three months following the end of period IV.
Tolerability
Serious adverse events were reported in four of the 32 patients in the clozapine group and in seven of the 28 patients in the placebo group during period II. In period III, the frequency of serious events was 27% (20% in the patients formerly in the clozapine group and 36% in those formerly on placebo). Overall, adverse events were more frequent or more severe in those patients receiving placebo first.
Withdrawals because of adverse events occurred with a similar frequency in both treatment groups (fig 1). In period II, the events leading to withdrawal were one neutropenia and one fracture in the clozapine group, and one hypotension and one syncope in the placebo group. In period III, the events leading to withdrawal were a sudden death, a death caused by aspiration pneumonia, one case of aggressiveness with delusions, and one case of confusion with delusions. This last patient died after the discontinuation of clozapine, as the result of a worsening of his general status.
Table 4 summarises adverse events occurring with a frequency > 10% during period II. Somnolence proved to be more frequent with clozapine than with placebo. One patient on clozapine (period II) experienced seizures. Transient neutropenia was seen in two clozapine treated patients (period II), leading to drug interruption in one patient, whereas normalisation was obtained despite clozapine continuation in the other. Worsening of parkinsonism was reported as an adverse event in 14 patients under clozapine (table 4), but only three of these patients experienced a decrease greater than 10% of the Schwab and England score. Of these three patients, the motor deterioration occurred at the time of withdrawal of a dopamine agonist drug for one and the two others received 50 mg/day of clozapine. This motor deterioration did not require treatment interruption.
Adverse events (⩾ 10%)
There was no worsening of motor symptoms in either group according to the UPDRS scores (table 5). Three patients in the clozapine group and one patient in the placebo group reached the criteria (decrease greater than 10% in the Schwab and England score) for interruption of clozapine dose escalation (p = 0.62).
Comparison of change scores of motor and cognitive functions in clozapine and placebo groups
No significant difference was seen between the two treatment groups for MMSE scores (table 4).
DISCUSSION
The efficacy of low dose clozapine on psychotic symptoms in patients with PD has been confirmed without ambiguity in this double blind study. The effects of clozapine compared with placebo were documented both on the CGI scale and the PANSS. Although recent attempts have been made to develop specific PD psychosis rating scales,15 most trials to date, including the US clozapine trial, continue to use rating scales validated for schizophrenic psychosis. The PANSS is a reference scale for the semiquantitative assessment of psychiatric symptoms in schizophrenia, in which positive syndrome items (hallucinations/delusions) have been selected as outcome variables of particular interest in the setting of dopaminometic induced psychosis.11 The positive PANSS subscore, and particularly the key items P1 (delusions) and P3 (hallucinations), were significantly improved by clozapine in our present study.
Appreciable improvement started a few days after the initiation of clozapine and increased continuously thereafter, reaching a maximum at three months, in accordance with previously published open trials.16 This early improvement, even at low doses, is of great clinical interest because of the distressing nature of the symptoms for patients and caregivers. The efficacy of clozapine was also evident during the open label period, in which patients formerly on placebo improved greatly once they received clozapine. The clinical relevance of the use of low dose clozapine was obvious because just over half of the patients had completely recovered from psychotic symptoms by the end of our study. Furthermore, more than one third of our patients could benefit from increased doses of levodopa or from the introduction of a dopaminergic agonist, without appreciable changes in clozapine doses and without recurrence of the psychiatric symptoms.
Previous open trials suggested that the effect of clozapine might rapidly vanish after drug withdrawal.17–19 An interesting aspect of our study was the systematic attempt to withdraw clozapine after complete remission of psychotic symptoms, which led to a relapse of psychotic symptoms in three quarters of the patients within one month. Thus, although it can be said that low dose clozapine has excellent efficacy on psychotic symptoms, the effect wears off once the treatment stops.
The overall frequency of adverse events in the clozapine group was not significantly greater than that of the placebo group. However somnolence, a well known dose related side effect, occurred more frequently on clozapine, despite a low starting dose. A possible deleterious effect of clozapine on motor function is one key point of the tolerability profile in PD. Some worsening of parkinsonism occurred in the clozapine treated patients. However, a reduction of more than 10% in the Schwab and England score was seen in only three patients. In these cases, the dose might have played a role. The few patients with motor worsening did not require treatment interruption and in most of the other cases, motor worsening proved to be transient and/or mild, and was not reflected by the Schwab and England or UPDRS scores. There were no significant changes between the clozapine and placebo groups in the UPDRS total score and subscores. This confirms that the improvement of psychosis did not occur at the expense of an unacceptable worsening of parkinsonism. Fewer data are available with the other atypical antipsychotics (olanzapine and risperidone), and more recent and wider experience has demonstrated a frequent and unacceptable worsening of parkinsonism with these drugs.20,21 As far as cognitive symptoms are concerned, the frequency of confusion and the decline in the MMSE scores did not differ between groups. Confusion was essentially limited to the placebo group, where baseline MMSE scores were slightly but significantly lower. Thus, the confusion that has been reported as an important side effect in open studies16 with clozapine could be related to the severity of cognitive impairment at baseline, rather than being a direct effect of clozapine. Serious adverse events were more frequent in the placebo group. Three deaths were reported in the open trial period, and ascribed to infections or primary cardiac events with uncertain relation to clozapine. Although sudden death is a well known adverse effect of neuroleptic drugs, it has also been reported in patients with PD.22 Moreover, a high risk of death was reported in these types of patients with advanced PD and psychosis.3 Two transient cases of neutropenia occurred on clozapine but no agranulocytosis, which is the major risk with clozapine. The incidence of agranulocytosis was 0.38% in a recent analysis of 99 502 US patients with schizophrenia and the risk might increase with age.14 To date, only two cases of agranulocytosis18,23 with a favourable outcome have been reported in PD. Thus, clozapine seems to be relatively safe in this patient group. The number of patients exposed to treatment was obviously not sufficient to establish conclusions in PD.
These results confirm the findings of the Parkinson study group,9 who used a similar methodology to that used here. However, more stringent criteria for the severity of psychosis were used in our study, and this could account for the higher mean clozapine dosage necessary to control psychotic symptoms (35.8 v 24.7 mg/day). In addition, the prevalence of cognitive decline differed slightly between both studies because MMSE scores below 20 were excluded in our study but not in the other trial. Given the similar efficacy of clozapine in both trials, pre-existing mental deterioration does not seem to be a major factor in treating patients with PD with clozapine
Conclusions
Our trial confirms that clozapine is an effective treatment of the psychotic complications induced by dopaminomimetic drugs, which does not significantly compromise motor and cognitive functions provided doses are less than 50 mg/day. Although is possible that a few patients would have benefited from doses higher than 100 mg/day, our study protocol did not permit higher doses, because the first double blind study of clozapine in parkinsonism showed worsening of motor function with higher doses.24 The efficacy of clozapine will allow an increase of antiparkinsonian treatments in selected patients. It appears that the treatment must continue after recovery, particularly if the symptoms were chronic before initiation. Tolerability was good in elderly, sometimes cognitively impaired, patients with advanced PD. Adverse events were mostly mild or moderate and could be controlled by slow dose titration. Therefore, clozapine is the only drug proved to be effective and well tolerated in the treatment of drug induced psychosis in PD as demonstrated by two double blind, placebo controlled trials. Because of the risk of agranulocytosis with clozapine, regular and long term blood cell counts are mandatory, and treatment should only be started if the usual therapeutic approach has failed. Although cost efficacy studies remain to be performed, one would expect them to be favourable because of the heavy cost of multiple hospitalisations and the institutionalisation rate linked to dopaminomimetic induced psychosis.
Acknowledgments
This work was sponsored by an educational research grant from Novartis Pharma France.
REFERENCES
Footnotes
-
Competing interests: PP has been reimbursed for expenses related to conferences, has received fees for speaking, and was once a consultant at the EMEA for Novartis. FT has been paid as an investigator in the present study and has been invited to several conferences by Novartis Pharma SA. OR and AD have been reimbursed for attending several conferences and received speaking fees from Novartis, the manufacturer of clozapine. JJP and IB are employees of Novartis, the sponsors of the study.
-
The French Clozapine Parkinson Study Group. Safety committee: J M Senard, Hôpital Purpan, Toulouse; V Lamarque, Novartis Pharma Rueil-Malmaison; and V Dev, Novartis Pharma Basle. Others members: Y Agid, Hôpital de la Salpêtrière, Paris; H Allain, CHU Rennes; M Borg, Hôpital Pasteur, Nice; E Broussolle, Hôpital Neurologique, Lyon; F Durif, Hôpital Gabriel Montpied, Clermont-Ferrand; C Geny, Centre Hospitalier Général, Pau; C Prunier-Levilon, CHU Bretonneau, Tours; S Sangla, Hôpital Delafontaine, Paris; M Vérin, CHU, Rennes; F Viallet, Hôpital Général, Aix-en-Provence; C Tranchant, Hôpital Civil, Strasbourg.