Article Text

Abstract
Objectives To study the safety and efficacy of vitamin D3 as an add on therapy to interferon β-1b (IFNB) in patients with multiple sclerosis (MS).
Methods 1 year, double blind, placebo controlled, randomised study in 66 MS patients. The primary outcomes were T2 burden of disease (BOD) on MRI scans, proportion of patients with serum levels of 25-hydroxyvitamin D (25(OH)D) ≥85 nmol/l or intact parathyroid hormone (PTH) ≤20 ng/l, and number of adverse events. Secondary outcomes were number of MRI enhancing T1 lesions and new T2 lesions, annual relapse rate, changes in the Expanded Disability Status Scale score, timed 25 foot walk test and timed 10 foot tandem walk tests.
Results Median change in BOD was 287 mm3 in the placebo group and 83 mm3 in the vitamin D group (p=0.105). Serum levels of 25(OH)D increased from a mean of 54 (range 19–82) nmol/l to 110 (range 67–163) nmol/l in the vitamin D group. 84% of patients reached a serum 25(OH)D level >85 nmol/l in the vitamin D group and 3% in the placebo group (p<0.0001). Patients in the vitamin D group showed fewer new T2 lesions (p=0.286) and a significantly lower number of T1 enhancing lesions (p=0.004), as well as a tendency to reduced disability accumulation (p=0.071) and to improved timed tandem walk (p=0.076). There were no significant differences in adverse events or in the annual relapse rate.
Conclusion Vitamin D3 add on treatment to IFNB reduces MRI disease activity in MS.
Trial registration number EudraCT number 2007-001958-99 and ClinicalTrialsGov number NCT01339676.
Statistics from Altmetric.com
Introduction
Finland is located between latitudes 60°N and 70°N and belongs to a high risk region for multiple sclerosis (MS), with a prevalence of 100–200 per 100 000 inhabitants in different areas.1 Diminished capacity of vitamin D to regulate immune responses as a consequence of low serum concentrations of 25-hydroxyvitamin D (25(OH)D) could be one environmental factor to explain the latitude association with MS prevalence.2–4 There is a growing body of evidence to support this vitamin D hypothesis in MS.5–10 The risk of MS is higher in individuals born in the spring and among women born to mothers with low milk or vitamin D intake during pregnancy.5 6 Intake of vitamin D from multivitamin supplements, high sun exposure in childhood or adolescence, and serum levels of vitamin D >100 nmol/l are all associated with a lower incidence of MS.7–10
Vitamin D also seems to regulate clinical disease activity in patients with established MS. MS patients have lower 25(OH)D levels during MS relapses than in remission.3 11 Prospective and retrospective studies have demonstrated a lower relapse risk with higher serum levels of 25(OH)D.12–14 Serum 25(OH)D levels correlate with disability in progressive forms of MS.14 15 Seasonal fluctuations in the numbers of gadolinium (Gd) enhancing MRI lesions in MS as well as an inverse correlation between brain MRI activity in MS patients and serum 25(OH)D at the population level have been described.16 17
In Finland, almost half of newly diagnosed MS patients have moderate to severe vitamin D deficiency during the winter.4 Most newly diagnosed Finnish MS patients start interferon β (IFNB) or glatiramer acetate therapy as soon as the McDonald diagnostic criteria for relapsing–remitting MS (RRMS) are fulfilled.18 19 The therapeutic efficacy of IFNB in MS is known to be only partial.20 Therefore, several trials investigating the efficacy of combination therapies have been initiated.20 21 Recently, a large multinational trial including 307 MS patients failed to show any benefit of 80 mg of simvastatin as an add on therapy to IFNB-1a.21 Nevertheless, vitamin D is considered to have tolerogenic immunological effects but is not known to influence lymphocyte trafficking, in contrast with IFNB.22–24 Therefore, the mechanism of action of vitamin D could be complementary to IFNB and lead to an additive clinical effect.
The specific objectives of this randomised, double blind, placebo controlled trial were to study: (a) whether a once weekly dose of 20 000 IU (500 μg) of vitamin D3 as an add on to IFNB-1b 250 μg subcutaneously every other day is safe and well tolerated and (b) to what extent it increases serum levels of 25(OH)D, and whether it has an effect on MRI and clinical variables (including T2 lesion load and new T2 or T1 Gd enhancing lesions, relapse rate and disability) in comparison with a group that received IFNB-1b and placebo capsules.
Methods
Patients
The Finnish Vitamin D Study (cholecalciferol as add on treatment to subcutaneously administered interferon-β 1b for the treatment of MS) was a double blind, randomised, parallel group, 1 year trial. The study protocol was approved by the ethics committee of Turku University and Turku University Hospital (28 August 2007, decision number 342) and the National Agency of Medicines, Helsinki, Finland (18 December 2007, decision number 220/2007). The study was undertaken in accordance with the Declaration of Helsinki and the European Medicines Agency Note for Guidance on Good Clinical Practice. Patients provided written informed consent before initiating study procedures.
Inclusion criteria
Inclusion criteria were: age 18–55 years; RRMS, according to the McDonald criteria,18 with IFNB-1b use for at least 1 month; no neutralising antibodies to IFNB, as measured by the indirect myxovirus A (MxA) test; Expanded Disability Status Scale (EDSS) score ≤5.0; using appropriate contraceptive methods (women of childbearing potential); and signed written informed consent.
Exclusion criteria
Exclusion criteria were: serum calcium >2.6 mmol/l; serum 25(OH)D >85 nmol/l; primary hyperparathyroidism; pregnancy or unwillingness to use contraception; alcohol or drug abuse; use of immunomodulatory therapy other than IFNB-1b; known allergy to cholecalciferol or peanuts; therapy with digitalis, calcitonin, vitamin D3 analogues or vitamin D; any condition predisposing to hypercalcaemia (such as any type of cancer), sarcoidosis, nephrolithiasis or renal insufficiency; significant hypertension (blood pressure <180/110 mm Hg); hyperthyroidism or hypothyroidism in the year before the study began; a history of kidney stones in the previous 5 years; cardiac insufficiency or significant cardiac dysrhythmia; unstable ischaemic heart disease; depression; and inability to perform serial MRI scans. Patients were recruited from the outpatient polyclinics of Turku, Helsinki, Tampere, Oulu and Kuopio University Hospitals and the Central Hospitals of Central Finland and Ostrobotnia.
Study product
Cholecalciferol (Dekristol) containing 20 mg of cholecalciferol, corresponding to 20 000 IU or 0.5 mg of vitamin D3, in arachis oil inside a refined gelatine capsule for once weekly peroral treatment or identically appearing matching placebo capsules (Swiss-Caps, Switzerland) were used. A private company (Joutsen Apteekki, Turku, Finland) organised the importing, packaging and labelling of the study product.
Randomisation and masking
Eligible patients were randomised 1:1 to treatment with either Dekristol or identically appearing matching placebo capsules. A separate randomisation was done for each centre using randomly permuted blocks. Randomisation was performed at 4Pharma Ltd using SAS for Windows software, V.8.2. The randomisation lists and all related documentation were stored in a secure place at 4Pharma Ltd. The investigator, patients, study nurses, radiologists and other persons directly involved in the conduct of the study were blinded to the treatment code, which was not opened until the database was locked. Laboratory values that could have revealed treatment allocation to the investigators (25(OH)D and intact parathyroid hormone (iPTH)) were sent directly to the data manager at 4Pharma Ltd. Laboratory analyses were performed at Turku University Hospital Central Laboratory, as described previously11 A commercially available assay, 25-hydroxyvitamin D 125I RIA Kit (DiaSorin Catalogue No 68100E, Stillwater, Minnesota, USA) was used for 25(OH)D measurements according to the instructions provided by the manufacturer. Two quality control samples were included in each assay series, and the specimens and controls were assayed in duplicate. The sensitivity of this assay is 4.0 nmol/l and the intra-assay coefficient of variation is <10%.
Procedures
There were six study visits over 12 months. At the screening visit, concurrent illnesses and concomitant medication were recorded. Physical examination, EDSS, height, weight, heart rate, blood pressure, ECG, timed 10 foot tandem walk (TTW10) and timed 25 foot walk (T25FW) tests were performed. Two attempts of both walk tests were recorded. Pregnancy test, MxA, serum calcium, creatinine, phosphate, alkaline phosphatase, albumin, magnesium, iPTH and 25(OH)D were measured. The baseline (=randomisation) visit was performed as soon as the laboratory results were available but not later than 4 weeks after the screening visit. MRI was done within 2 weeks before or at the randomisation visit. Eligible patients were randomised to cholecalciferol or placebo capsules. A bottle of 26 capsules of study medication was given to the patients and the investigator instructed the patients in the weekly administration of the study medication. Serum levels of calcium, creatinine and phosphate were measured at 1, 2, 3, 6, 9 and 12 months. Additionally, plasma levels of alkaline phosphatase, albumin, magnesium, iPTH and 25(OH)D were measured at 6 and 12 months, ECG at 2, 6 and 12 months and MxA at 12 months. EDSS and timed walk tests were measured again at 12 months. At the 6 month visit, the number of returned capsules were counted and a new bottle of 26 capsules of study medication was given to the patient. Patients contacted the study centres for unscheduled visits within 7 days of a relapse onset. At the unscheduled visit, an EDSS was performed and the investigator defined whether a relapse had occurred. At the judgement of the treating neurologist, relapses were treated with methylprednisolone 1 g daily for 3 days. Adverse events were assessed at each visit. The primary endpoints were T2 burden of disease (BOD), proportion of patients with serum levels of 25(OH)D ≥85 nmol/l or iPTH ≤20 ng/l at 6 and 12 months, and safety and tolerability (number of adverse events). Secondary endpoints included change in EDSS, relapse rate, time to first relapse, change in TTW10 and T25FW, number of T1 enhancing lesions, T1 enhancing lesion volume (mm3), number of new or enlarging T2/PD lesions and MRI activity (defined as the presence of Gd and/or new/enlarging T2 lesions).
MRI acquisition and analysis
Patients underwent a standardised MRI study in each centre using a 1.5 T scanner. MRI was performed within 2 weeks before or at the randomisation visit and within 2 weeks before or at the final visit. Before a site was accepted into the trial, a dummy run MRI was performed. The scanning protocol included a dual echo T2/PD and a post-contrast T1 weighted sequence covering the whole brain in transverse imaging plane with continuous 3 mm slices. Central analyses were performed at the Neuroimaging Research Unit, Vita-Salute University, Milan, Italy, and included quantification of the total number of Gd enhancing lesions, number of new/enlarging T2/PD lesions and new Gd enhancing lesions, T2 lesion volume (BOD) (mm3) and T1 enhancing lesion volume (mm3). Lesion volumes were quantified by experienced observers using a local thresholding segmentation technique (Jim 5, Xinapse Systems Ltd, Northants, UK).
Statistical analysis
SAS V.9.2 was used for all analyses. The sample size calculation was performed such that with 40 patients in each treatment arm a difference of 1000 mm3 in MRI T2 BOD (SD 1700) and a difference of 30% in the proportion of patients with 25(OH)D ≥85 nmol/l would be detected (two sided χ2 test, α=0.05, power=80%). Logistic regression was used to analyse vitamin D status by visit and to analyse MRI activity. Non-parametric rank analysis of covariance (ANCOVA) was used in analysing MRI T2 BOD and EDSS at 12 months with baseline values as covariates, controlling for centre. ANCOVA was used to analyse the change in TTW10 or T25W (after log transformation), with TTW10 or T25W at baseline and centre as covariates. Numbers of Gd enhancing lesions on T1 and new/enlarging lesions on T2 scans were analysed using a generalised linear mixed model based on Poisson distribution. T1 enhancing lesion volume was analysed using Fisher's exact test at each time point. Correlations between EDSS and MRI T2 BOD were calculated by Spearman's rank correlation coefficient. A p value <0.05 was considered statistically significant.
Role of the funding source
This research was investigator initiated and investigator driven, with Bayer providing an unrestricted grant. The principal investigator (MHS-H) wrote the protocol, supervised the study and interpreted the results. Data management, statistical analyses and monitoring were done by independent contract research organisations (4Pharma Ltd, Turku, Finland and Premedic Clinical Research Ltd, Turku Finland). The sponsor did not have any role in the design of the study, data collection, statistical analysis or interpretation of the data. The principal investigator had full access to all of the data in the study after the database lock.
Results
A flowchart of the study is shown in figure 1. Patient demographics and clinical characteristics at baseline are shown in table 1. MRI characteristics at baseline are shown in table 2. The first patient was screened in March 2008 and randomised in April 2008. The last patient was randomised in May 2010 and completed the study in May 2011. The database was locked in August 2011.

Flowchart of the patients in the study.
Patient demographics and clinical characteristics at study entry
MRI parameters in vitamin D and placebo treated patients at baseline and at 12 months
Primary outcomes
Serum vitamin D and PTH levels
As expected, the percentage of patients with vitamin D status of 25(OH)D >85 nmol/l was significantly higher in the vitamin D treated group compared with placebo group at both 6 and 12 months (6 months: 76% vs 3%, p<0.001; 12 months: 84% vs 3%, p<0.001). Mean serum 25(OH)D was 54 (range 19–82) nmol/l at baseline and 110 (67–163) nmol/l at 12 months in the vitamin D treated group. In the placebo group, mean serum 25(OH)D was 56 (range 16–81) nmol/l at baseline and 50 (17–94) nmol/l at 12 months. The difference between the vitamin D and placebo groups was statistically significant (p<0.0001). Serum vitamin D values at study baseline, and at 6 and 12 months are shown in figure 2.
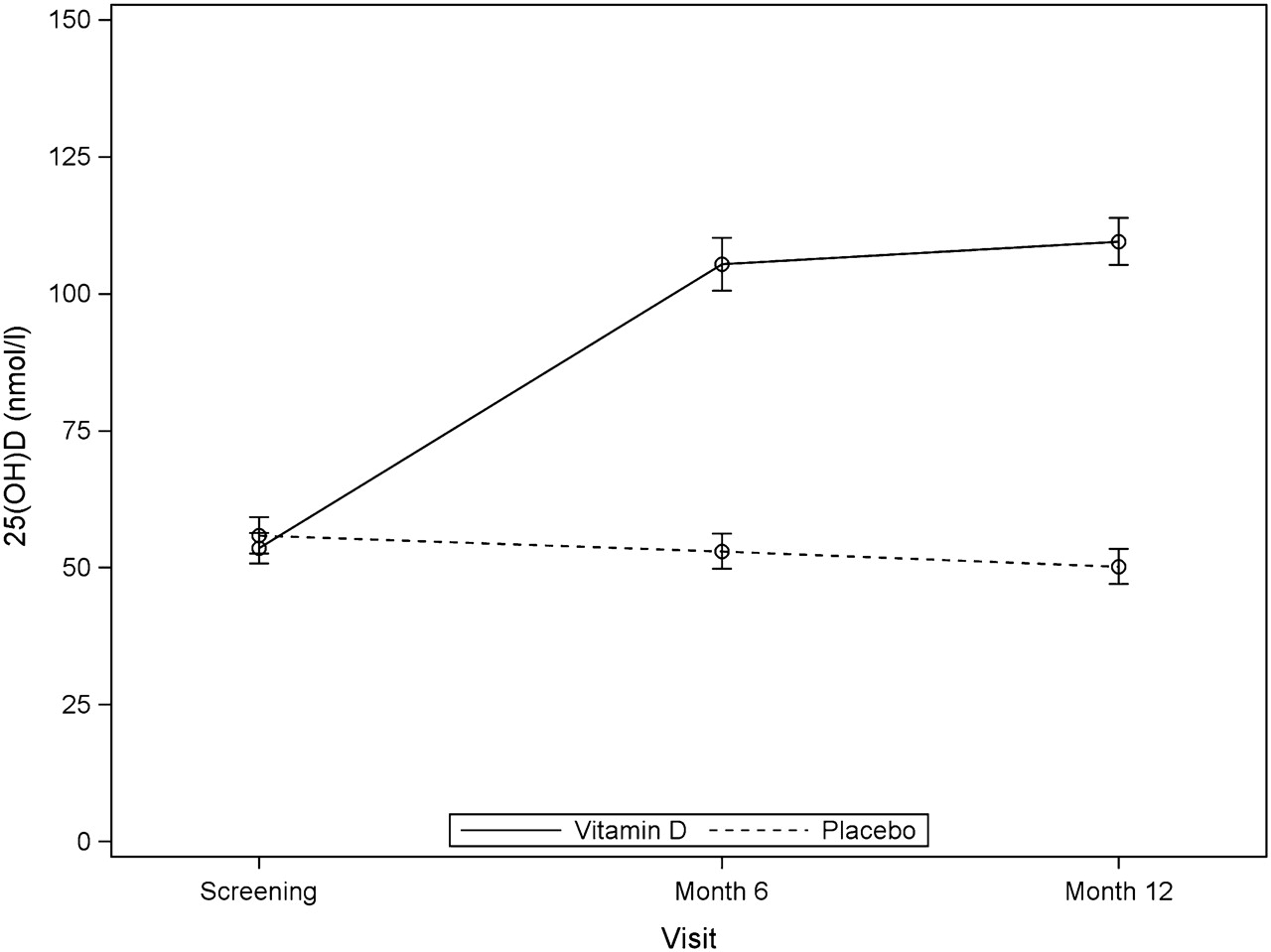
Serum levels of 25-hydroxyvitamin D (25(OH)D) at screening, and at the month 6 and month 12 visits in the vitamin D treated and placebo treated patients. Laboratory reference range for 25(OH)D is 40–375 nmol/l. Data are mean (SE). Serum levels of 25(OH)D were statistically significantly different between the vitamin D treated and placebo patients (p<0.001).
PTH suppression to a level <20 pg/ml was not obtained in either treatment arm (see supplementary figure 1A; available online only). In the vitamin D treated patients, median iPTH at baseline was 37 (range 1–77) pg/ml and at 12 months 36 (20–67) pg/ml. In the placebo group, median serum level of iPTH at baseline was 41 (range 15–69) pg/ml and at 12 months 42 (18–125) pg/ml.
Safety and tolerability
There was no hypercalcaemia in vitamin D treated patients (supplementary figure 1B, available online only) or significant differences between the treatment arms in any of the other clinical chemistry parameters studied (data not shown). No dose adjustments were necessary. Lack of MxA response (MxA <50 μg/l) was detected in three patients in both treatment arms at 12 months. Diarrhoea was a side effect in five patients in the vitamin D group and in two patients in the placebo group and fever was noted in five patients in the placebo group and in two patients in the vitamin D group. All other adverse events occurred in a similar number of patients in both groups. There was one serious adverse event in the vitamin D group (erysipelas in the interferon injection site treated with intravenous antibiotics in hospital) and two in the placebo group (elective hip surgery and elbow fracture).
MRI T2 burden of disease
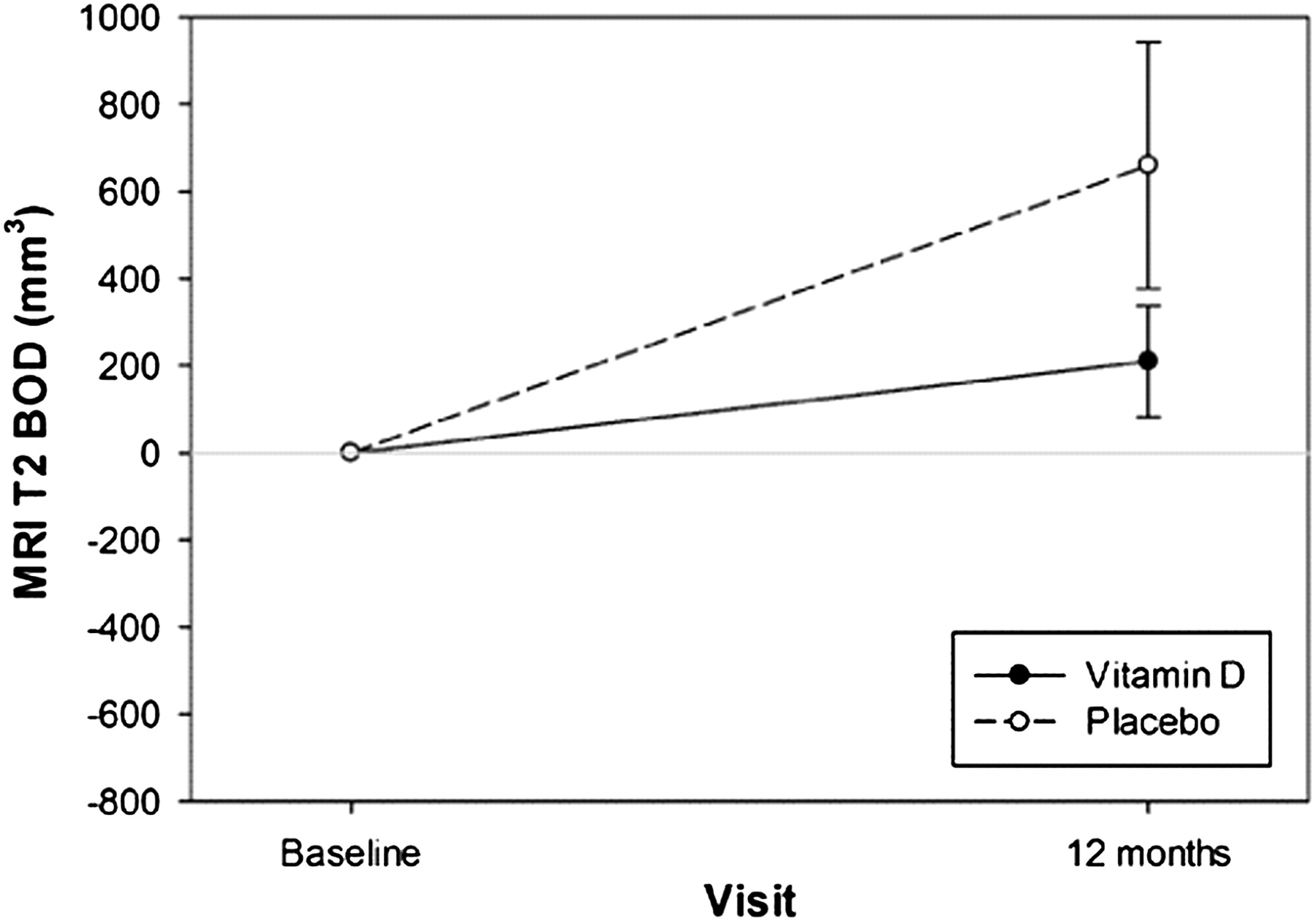
MRI T2 BOD, the primary MRI outcome measure, increased more in the placebo patients (median change 287 mm3) compared with the vitamin D treated patients (median change 83 mm3) but the difference between the treatment groups was not statistically significant (p=0.105) (figure 3, table 2). When patients with a lack of MxA response were excluded from the analysis, the p value became close to significant (0.055).

{kind=link}
{kind=link}
{kind=link}
Change in MRI T2 burden of disease (BOD) from baseline to month 12 in the vitamin D treated and placebo treated patients. Data are mean (SE) of 34 patients in the vitamin D group and 32 patients in the placebo group at baseline and 32 patients in the vitamin D group and 30 patients in the placebo group at 12 months. The p value for difference between vitamin D and placebo is 0.105 (trend).
Secondary outcomes
Total number of Gd enhancing T1 lesions, new/enlarging T2 lesions, Gd enhancing lesion volume and MRI activity
The total number of T1 Gd enhancing lesions decreased significantly in both groups (p=0.002) but this change was significantly higher in the vitamin D group (p=0.004) (table 2). The number of new/enlarging T2 lesions at 12 months was higher in the placebo group but the difference was not statistically significant (p=0.286) (table 2). The volume of Gd enhancing lesions at 12 months did not differ between the two groups (table 2). The percentage of patients with MRI activity at 12 months was lower in vitamin D treated patients but the difference was not statistically significant (p=0.322).
Relapses, EDSS, and T25FW and TTW10
The annual relapse rate decreased during the study in both treatment arms (table 3). There was no significant difference in the time to first relapse between groups (HR=1.12, 95% CI 0.41 to 3.1) (supplementary figure 2, available online only). The proportion of relapses treated with methylprednisolone was similar in both treatment arms (4/9 relapses in both groups). The mean EDSS score decreased in the vitamin D group and remained the same in the placebo group during the study (p=0.071) (table 3). In the whole group of patients, EDSS was significantly correlated with T2 BOD (supplementary figure 3, available online only). When the walk test results at 12 months were compared with baseline values, there was a statistically significant decrease in TTW10 in the vitamin D group (p=0.001) and a non-significant decrease in the placebo group (p=0.291). The difference between the groups between baseline and 12 months was not statistically significant (p=0.076) (table 3). There was no statistically significant differences between the treatment groups in T25FW (p=0.907) (table 3).
Clinical outcomes in vitamin D treated and placebo patients
Discussion
Our study is the first randomised, double blind, placebo controlled trial examining the effects of vitamin D3 in MS. In a recent Cochrane review, randomised and quasi-randomised controlled trials comparing vitamin D with placebo or any other treatment for MS were considered.25 Only one open label, randomised, prospective, controlled trial that treated 25 Canadian MS patients with escalating doses of vitamin D compared with 24 controls met the selection criteria.26 In the study published by Burton et al, a high dose (10 000 IU/day) of vitamin D3 was found to be safe with evidence of immunomodulatory effects.26 The trial was not powered or blinded to properly address clinical outcomes. Their control group was allowed to take up to 4000 IU/day of vitamin D and supplementary calcium if desired. However, clinical outcomes appeared to favour the high dose vitamin D group. Our trial was properly blinded and the placebo patients did not take any vitamin D, but due to the small sample size, the trial was not powered to address clinical outcomes. The dose of vitamin D in our study was lower than in the Canadian trial (20 000 IU/week). Similar to the Canadian trial, a trend favouring the vitamin D group in EDSS scores was seen in our trial. The value of our EDSS finding was strengthened by a statistical trend in the primary outcome measure, MRI BOD, and a significant correlation between the EDSS values and MRI BOD in our patients.
Recently, a study based on patient reported previous sun exposure, vitamin D intake and age at disability milestones from 219 veterans with progressive forms of MS in the Multiple Sclerosis Surveillance Registry was published.27 The study found that a low average sun exposure before disease onset increased the risk to disease progression whereas use of cod liver oil during childhood delayed progression.27 We did not measure brain atrophy and followed disability accumulation for only 1 year. Therefore, alternative explanations for our results favouring the vitamin D group in disability accumulation and timed tandem walk other than reduced neurodegeneration have to be considered. Doses of 17.5–25 μg/day (700–1000 IU/day) of vitamin D have been shown to significantly lower the risk of fall in older adults.28 The minimal serum level of 25(OH)D that was needed to obtain benefit for balance was 60 nmol/l.28 An effect of vitamin D on muscle performance and balance could account for the tendency towards improved timed tandem walk and EDSS scores that we observed in the vitamin D3 treated MS patients in comparison with the placebo treated patients.
In a previous open trial involving 12 patients supplemented with 1000 μg/day of vitamin D for 28 weeks, a decline in the mean number of Gd enhancing brain MRI lesions per patient was seen.29 The EPIC cohort study evaluating 469 subjects with a clinically isolated syndrome or RRMS found that higher vitamin D serum levels were associated with the development of fewer T2 and Gd enhancing brain MRI lesions.30 Our results are in line with these findings. In contrast, in a newly published Australian randomised controlled trial in 23 people with clinically active RRMS taking 1000 IU of vitamin D2 per day, no therapeutic advantage from additional high dose vitamin D2 of 6000 IU/day was found. However, the trial was limited by a small and selected patient sample. Nineteen of the patients were either receiving glatiramer acetate or IFN therapy and three patients withdrew, making the ultimate number of comparable patients in each treatment arm very small.31
In the study by Burton et al, decreased T cell proliferative responses were observed in a significantly larger proportion of those patients who achieved 25(OH)D levels ≥100 nmol/l.26 In our study, a modest dose of 20 000 IU/week of cholecalciferol increased serum 25(OH)D levels to a mean of 110 nmol/l. To date we have not performed immunological analyses in our patients but the significant decrease in Gd enhancing T1 lesions suggests a reduction in brain inflammatory activity at the vitamin D dose of 20 000 IU/week that we used in this study.
After the publication of the Cochrane review, a double blind, placebo controlled, randomised, 96 week trial in 68 Norwegian MS patients and a 6 month randomised controlled trial with 62 Iranian MS patients were published.32 33 In the study performed in Norway, bone mineral density was the primary outcome measure. The vitamin D3 product and placebo were identical to our trial. Baseline 25(OH)D levels were similar (approximately 55 nmol/l in the Norwegian study as in ours) and a mean serum 25(OH)D concentration of 123 nmol/l was achieved with the 20 000 IU/week dose of cholecalciferol and the low dose vitamin D supplement that they used at study baseline and were allowed to continue. In the Iranian trial, patients were randomised to once monthly intramuscular 300 000 IU vitamin D injections (n=28) or placebo intramuscular injections (n=34), and EDSS, mean number of brain Gd enhancing lesions, relapses and T cell function were studied at baseline and at 6 months. No significant differences were found in clinical or MRI parameters in the Iranian trial but lymphocyte proliferation was decreased in the treated patients. Serum levels of 25(OH)D of approximately 150 nmol/l were achieved in their study. Adverse effects with vitamin D supplemented at a higher than the generally recommended dose of vitamin D were not shown in any of these trials. In our trial, all patients used IFNB-1b and showed a decreased annual relapse rate from 0.51 to 0.26 and 0.28 in the placebo and vitamin D groups, respectively. Conceivably, vitamin D did not exert any additional effect on relapse rate.
In conclusion, this randomised, double blind, placebo controlled trial with 20 000 IU/week of vitamin D3 in RRMS patients using IFNB showed a statistically significant reduction in the number of T1 enhancing lesions and trends in MRI BOD and EDSS. The study was not powered for the clinical endpoints. Larger randomised controlled trials with longer follow-up than 1 year are needed to confirm the promising MRI results and to fully address clinical outcomes.
Acknowledgments
The authors thank all of the patients who participated in the study. The study nurses Jaana Nummela (Turku), Pirjo Hagman and Sirpa Esa (Helsinki), Raija Paalavuo (Tampere), Aino Mäkitalo and Aija Kallio (Jyväskylä), Maarit S Nappa and Tuula Ahola (Oulu), Johanna Viitaniemi (Seinäjoki), and Tarja Lappalainen and Helena Mäkelä (Kuopio) performed the timed walking tests. Turku University Hospital Central Laboratory analysed the blood samples. The authors thank Tuula Laukkanen for skilled assistance and support with the laboratory analyses throughout the study, and also Ritva Lindgren for monitoring and Jari Turunen, Elina Tenhunen and Vesa Mäkiaho for data management. The authors also acknowledge the following: Ostrobothnia: Susanna Hintikka (acquisition of data), Keijo Koivisto (administrative, technical and material support); Oulu: Mauri Reunanen (administrative, technical and material support); Kuopio: Merja Hallikainen, Lasse Nieminen (acquisition of data), Tuula Pirttilä (administrative, technical and material support, study concept and design).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
See Editorial commentary, p 473
Linked article 302422.
Funding The study was funded by an unrestricted grant from Bayer. MS-H was funded by the Finnish MS Foundation and by the Finnish Brain Foundation.
Competing interests None.
Ethics approval The study was approved by Turku University Ethics Committee and the Finnish Agency of Medicines.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary