Article Text

Abstract
Background and objective Historically, the diagnosis of sporadic inclusion body myositis (IBM) has required the demonstration of the presence of a number of histopathological findings on muscle biopsy—namely, rimmed vacuoles, an inflammatory infiltrate with invasion of non-necrotic muscle fibres (partial invasion) and amyloid or 15–18 nm tubulofilamentous inclusions (Griggs criteria). However, biopsies of many patients with clinically typical IBM do not show all of these histopathological findings, at least at presentation. We compared the clinical features at presentation and during the course of disease in 67 patients with histopathologically diagnosed IBM and clinically diagnosed IBM seen within a single UK specialist muscle centre.
Methods and results At presentation, using clinically focused diagnostic criteria (European Neuromuscular Centre (ENMC) 2011), a diagnosis of IBM was made in 88% of patients whereas 76% fulfilled the 1997 ENMC criteria and only 27% satisfied the histopathologically focused Griggs criteria. There were no differences in clinical features or outcomes between clinically and histopathologically diagnosed patients, but patients lacking the classical histopathological finding of rimmed vacuoles were younger, suggesting that rimmed vacuoles may be a later feature of the disease.
Conclusions These findings have important implications for diagnosis and future studies or trials in IBM as adherence to histopathologically focused diagnostic criteria will exclude large numbers of patients with IBM. Importantly, those excluded may be at an earlier stage of the disease and more amenable to treatment.
- Incl Body Myositis
- Muscle Disease
Statistics from Altmetric.com
Introduction
Historically, sporadic inclusion body myositis (IBM) has been classified as an idiopathic inflammatory myopathy, along with dermatomyositis and polymyositis (PM). However, clinically, there are rather more differences than similarities between IBM and the other two entities, particularly early involvement of finger flexor and quadriceps muscles, often asymmetric weakness and lack of response to immunosuppression in IBM.1 ,2 Weakness of the quadriceps often causes patients to present with falls, and finger flexor weakness leads to profound problems with activities of daily living. The clinical course is invariably slowly progressive with most patients eventually requiring mobility aids and many becoming severely disabled. Given the generally late onset, it only infrequently shortens life expectancy.2
Whether IBM is primarily an inflammatory or degenerative disease is debated, with a consistently poor response to immunosuppressive medication favouring the latter. Largely because of our incomplete knowledge of the cause of IBM, there is currently no single gold standard diagnostic test and consequently a number of diagnostic criteria have been proposed, most of which have emphasised histopathological findings.3–7 Probably the most extensively used and quoted diagnostic criteria are referred to as the Griggs criteria5; these require the presence in a muscle biopsy of an inflammatory infiltrate, inflammatory cell invasion of intact fibres (partial invasion), rimmed vacuoles and the presence of either amyloid deposits or 15–18 nm tubulofilamentous inclusions, shown on electron microscopy (EM), for a definite diagnosis of IBM to be made. A diagnosis of possible IBM can be made if the first three histopathological features are present in addition to specific clinical features.
Although the histopathological features in combination are thought to be highly specific to IBM, their sensitivity is increasingly being questioned. Since the delineation of IBM as a separate entity from PM in the early 1970s, it has been realised that most patients have a very characteristic clinical picture in terms of the mode of presentation, pattern of weakness and progression. However, patients with the characteristic clinical features may lack the classical histopathological findings, even despite repeated biopsy.1 ,8 This is compounded by the fact that in some laboratories, staining for amyloid and EM may not be available. The clinical findings are thought to be highly specific and sensitive for IBM and in experienced hands allow a diagnosis of IBM to be made with a supportive, if not diagnostic, biopsy.1 ,9 To enable inclusion of such patients in research studies, modified diagnostic criteria10 ,11 have been suggested, and consensus criteria were agreed at a recent European Neuromuscular Centre (ENMC) workshop in 2011 (table 1).12 This is particularly important given the suspicion that patients lacking the classical histopathological features of IBM may represent an earlier stage in the disease process and are, therefore, potentially more amenable to therapeutic interventions. Several publications have included patients lacking the typical histopathological features1–3 ,8 ,13–16; two of these indicated that the clinical features at presentation may be sensitive and specific for the diagnosis.1 ,13 However, the long term outcome in these patients is uncertain and diagnosis currently is predominantly based on personal experience.
Proposed 2011 European Neuromuscular Centre diagnostic criteria for inclusion body myositis
Here we describe and compare the clinical assessment and histopathological findings at presentation and investigate disease progression in 67 patients with IBM.
Methods
Case definition
All patients with a diagnosis of IBM seen within the muscle service at the Radcliffe Infirmary/John Radcliffe Hospital, Oxford, UK (between 1982 and 2011) and whose muscle biopsy and clinical data were available from their first presentation were included. The following data were collected prospectively at hospital appointments and collated retrospectively: date of birth, gender, ethnicity, past medical history, nature and onset of the disease, date of first symptoms, date IBM diagnosed, alternative initial diagnoses, physical examination at presentation and last follow-up, laboratory investigations at presentation, details of immunosuppressive treatment given for their presenting muscle problem, date requiring walking aid (walking stick or frame) and wheelchair. Disease onset was approximated to the year the patient recalled their first symptoms. Delay to diagnosis was taken as the time from symptom onset until the diagnosis of IBM was confirmed either by muscle biopsy or specialist clinical opinion.
Muscle biopsies
Muscle biopsies were reviewed by a single investigator (SB) blinded to the clinical details. Mitochondrial changes were assessed on combined cytochrome oxidase/succinate dehydrogenase staining which was available for 56 cases. Major histocompatibility complex class I (MHC class I) stained sections were available for review in 50 cases. Definitions of histopathological features are summarised in the online supplementary table S1.
Diagnostic criteria
Patients were classified according to the histopathological Griggs criteria, 1997 ENMC criteria and the clinical 2011 ENMC criteria. To enable comparisons between the histopathologically and clinically diagnosed groups, Griggs definite and Griggs possible IBM were combined into one group, as were 2011 ENMC clinically defined and probable IBM.
Statistical analysis
Statistical analysis was performed using GraphPad PRISM V.5. Continuous variables are presented as medians (IQR). Groups were compared using the Mann–Whitney U test. Categorical variables were analysed using Fisher's exact or χ2 test. Kaplan–Meier survival curves were used to estimate time to disability endpoints. Survival curves were compared using Cox proportional hazards model. Statistical significance was set at p<0.05. Patients who had received immunosuppressive treatment for conditions other than IBM were excluded from comparisons between treated and untreated patients.
Results
Demographic data
Sixty-seven patients were included; 66 (99%) patients were Caucasian and one was Asian. Forty-six patients (69%) were males (table 2). Median disease duration at the last follow-up was 9 years (6–14 years). Median age at symptom onset was 62 years (55–70) and median age at muscle biopsy was 67 years (61–75); this was significantly lower in patients without rimmed vacuoles on muscle biopsy (66 vs 74 years; p=0.04). Median delay to diagnosis was 62 months (34–75) and 26 (39%) received at least one alternative diagnosis before being diagnosed with IBM, most commonly PM.
Demographic characteristics
Presentation and distribution of weakness
The initial symptoms in all patients were falls and/or weakness, including dysphagia. Fifty-five (82%) patients reported lower limb weakness, predominantly proximal, as their initial symptom, five (8%) weakness of grip and one (2%) dysphagia. The remaining six patients’ initial symptoms were a combination of upper and lower limb weakness (6%) or dysphagia and lower limb weakness (3%). The three most frequently affected muscle groups at presentation were knee extensors (87%), finger flexors (79%) and ankle dorsiflexors (66%) (table 3). Knee extensor weakness was greater than or equal to hip flexor weakness in 55 (82%) patients and finger flexor strength was less than shoulder abductor strength in 43 (64%) patients. At presentation, 34 (51%) patients had finger flexor strength less than shoulder abductor strength and knee extensor weakness greater than or equal to hip flexor weakness. Over the period of follow-up, 55 (82%) patients developed this characteristic pattern of weakness. There were no differences in the presentation or distribution of muscle weakness between patients with and without rimmed vacuoles or between male and female patients.
Clinical examination at presentation
Muscle biopsy
Endomysial inflammation was present in 62 (93%) muscle biopsies, partial invasion in 56 (84%), rimmed vacuoles in 29 (43%) and all three in only 26 (39%) (table 4). EM is not routinely performed in our centre; in the 22 cases in which it was performed, 14 (64%) were positive for 15–18 nm tubulofilamentous inclusions. In 13 of those 14 cases, rimmed vacuoles were present. Sarcolemmal expression of MHC class I was diffusely increased in 46 (92%) and cytochrome oxidase negative/succinate dehydrogenase positive fibres were observed in 41 (73%) cases. Nine patients who lacked all three light microscopic features required by the Griggs criteria on their initial muscle biopsy had a second biopsy performed; this revealed all three histopathological features in four cases. Ten biopsies which lacked rimmed vacuoles on the initial routine sections had multiple additional sections cut and stained; rimmed vacuoles were discovered in only one case.
Creatine kinase and muscle biopsy findings
Treatment and disability
Eight (12%) patients received a combination of prednisolone with azathioprine or methotrexate. Median duration of treatment was 60 months (9–120) and five patients reported an initial benefit. However, objective improvement was observed in only two cases and was of short duration. Immunosuppression was withdrawn in seven cases due to lack of efficacy and in one because of side effects.
Forty-two (63%) patients required a walking aid by the last follow-up and a further 19 (28%) required a wheelchair. Median disease duration from symptom onset to requiring a walking aid or wheelchair was 96 and 228 months, respectively. Age at disease onset influenced disability; patients presenting at an older age required a walking aid sooner than younger patients (p=0.001). Gender, diagnostic criteria, histopathological features and immunosuppressive treatment did not influence the time to requiring a walking aid or wheelchair (figure 1A–F).
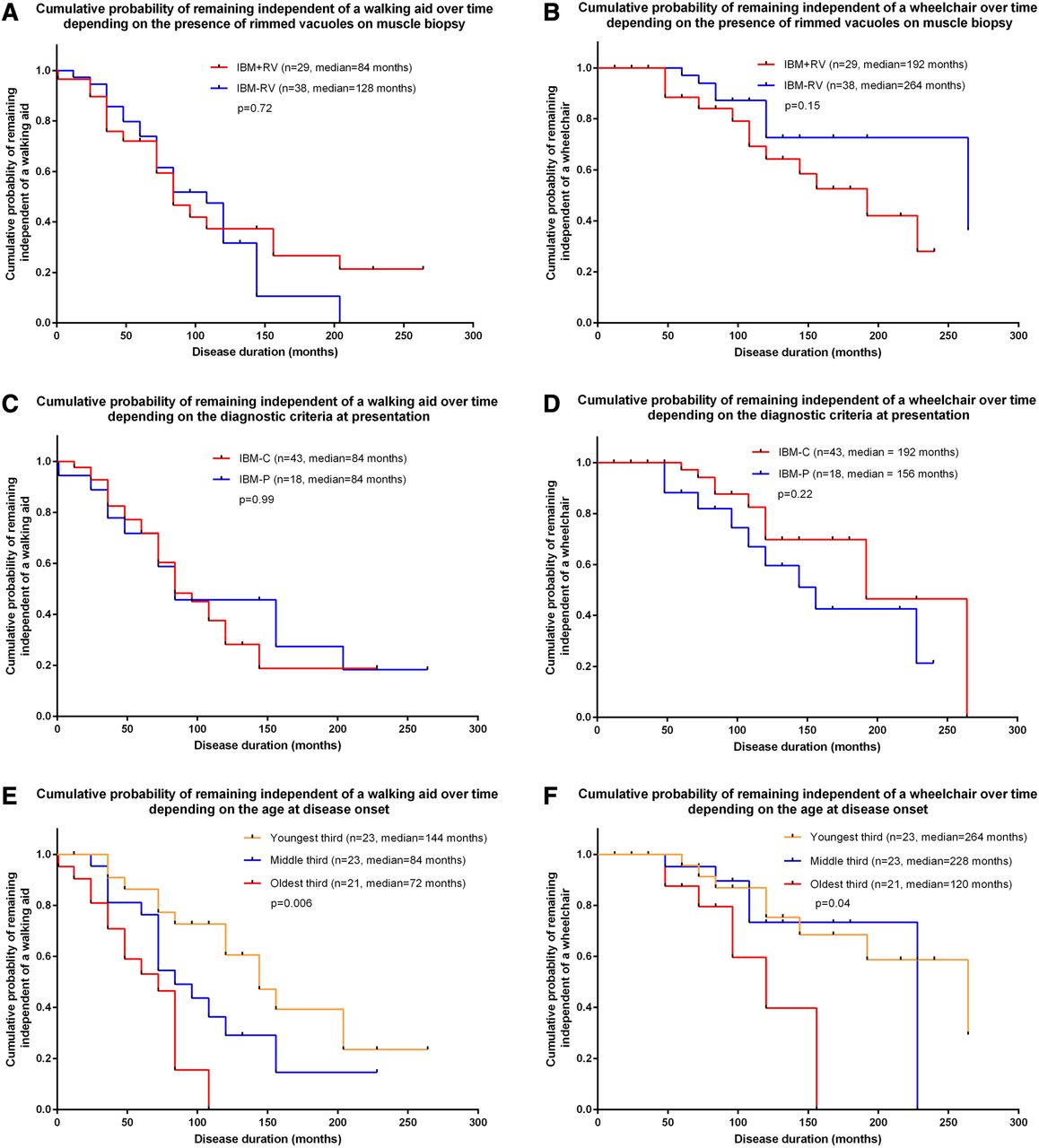
{kind=link}
Kaplan–Meier curves showing the cumulative probability over time of requiring a mobility aid (walking aid and wheelchair) depending on the presence of rimmed vacuoles on muscle biopsy (A, B), histopathologically (Griggs definite and Griggs possible) or clinically diagnosed inclusion body myositis (IBM) (2011 European Neuromuscular Centre (ENMC) clinically defined and 2011 ENMC probable) (C, D) and age at disease onset (E, F). Time to requiring a mobility aid was not influenced by histopathological features or diagnostic criteria (A–D). Age at disease onset affected the time to requiring a mobility aid; the time to require a walking aid (p=0.006) (E) and wheelchair (p=0.04) (F) was significantly longer in patients with their first symptoms at a younger age. Multivariate analysis revealed that age, calculated as a continuous variable, influenced time to requiring a walking aid (p=0.001; HR=1.07; 95% CI 1.03 to 1.11) but not a wheelchair (p=0.13; HR=1.05; 95% CI 0.99 to 1.12). IBM+RV, IBM with rimmed vacuoles on muscle biopsy; IBM-RV, IBM without rimmed vacuoles on muscle biopsy; IBM-C, clinically diagnosed IBM (2011 ENMC clinically defined and 2011 ENMC probable) excluding histopathologically diagnosed IBM; IBM-P, histopathologically diagnosed IBM using the Griggs criteria (Griggs definite and Griggs possible).
Diagnostic criteria
Fifty-nine (88%) patients fulfilled the 2011 ENMC criteria at presentation (11 (16%) clinicopathologically defined, 26 (39%) clinically defined and 22 (33%) probable) while only 18 (27%) fulfilled the Griggs criteria and 51 (76%) patients satisfied the 1997 ENMC criteria (definite 23 (34%), probable 27 (40%) and possible one (1%)). Sixteen (89%) patients fulfilling the Griggs criteria also fulfilled the 2011 ENMC criteria. Failure to satisfy the Griggs criteria was due to incompletely meeting the biopsy criteria in 15 (22%) cases, clinical criteria in eight (12%) and both in 26 (39%) cases. Eight patients did not fulfil the 2011 ENMC criteria; this was due to age at onset being <45 years in five (7%), failure to meet the examination criteria in two (3%) and the biopsy criteria in one (1%). There were no differences in age at presentation, gender, presenting symptoms, clinical examination findings, presence of concomitant diseases, creatine kinase (CK), biopsy site or histopathological findings (excluding rimmed vacuoles and tubulofilamentous inclusions), or immunosuppressive therapy received between clinically and histopathologically diagnosed patients.
Discussion
IBM was originally identified as a ‘steroid resistant’ form of PM but on further assessment was noted to have specific histopathological and clinical features.17 ,18 This led to the development of the histopathologically focused Griggs diagnostic criteria. Individually, the histopathological features of the Griggs criteria are non-specific.19–22 However, when present together they are considered diagnostic of IBM. Clinical experience over the intervening 40 years has supported early observations that IBM is associated with a very characteristic pattern of muscle involvement with predilection for the long finger flexors and quadriceps, and occasionally the muscles associated with swallowing.1 ,23 In our experience, patients with this clinical presentation, who do not fulfil the Griggs histopathological criteria, are at least as common as those who do. Other studies have described clinical or histopathological findings in patients not fulfilling Griggs criteria but detailed clinical data are lacking in most and long term outcome data on clinically diagnosed patients are lacking.1–3 ,8 ,13–16 Two of these studies included clinical examination findings in agreement with our current findings.1 ,13
We have characterised the clinical and histopathological features of patients with IBM and found that the clinical features are more sensitive than the histopathological findings in establishing a diagnosis.
The demographics, clinical features, speed of disease progression and response to immunosuppressive therapy in our cohort were similar to previous studies.1 ,9 ,13 ,24 Although dysphagia affects a substantial proportion of patients with IBM at some stage during the disease course,25 we found it to be uncommon as an initial symptom, although a proportion had developed mild dysphagia by the time patients were seen within our specialist service. We found no differences in clinical characteristics or disease progression between clinically and histopathologically diagnosed patients. In addition, cases with and without rimmed vacuoles, sometimes considered a sine qua non for diagnosis, were clinically identical except for age at biopsy; specifically, there was no difference in the pattern of weakness or disease progression. These findings confirm our long term observation that we are dealing with a clinically homogenous disorder, irrespective of whether patients are diagnosed on the basis of clinical or histopathological criteria and that IBM can be accurately and reliably diagnosed without fulfilling all of the Griggs histopathological features. In concordance with previous studies, we found that age at symptom onset predicted disease progression.24 ,26 Older onset patients had a more rapid decline and required walking aids earlier. Suggested reasons for this have included age related sarcopenia and the higher frequency of comorbidities in older patients.2 ,24 Although our data appeared to show that male patients reached disability endpoints more rapidly than female patients, this was an effect of age.
The absence of the Griggs histopathological features in a substantial proportion of our cohort may be due to the vagaries of sampling, or alternatively these features may indicate an advanced stage of disease progression and are therefore absent on the initial assessment.1 ,3 Because of the insidious nature of IBM, it is difficult to accurately identify disease onset or duration prior to diagnosis; however, we found that patients without rimmed vacuoles on muscle biopsy were younger at the time of biopsy than patients with them. This observation lends support to the notion that some of the histopathological features are absent early in the disease course and, therefore, are of limited diagnostic potential at the time of initial presentation. Differences in the frequency of partial invasion and mitochondrial changes have previously been described between cases with and without rimmed vacuoles.13 ,15 In agreement with a previous publication,13 we found the absence of both partial invasion and mitochondrial abnormalities to be strong evidence against the diagnosis of IBM. Divergent observations to ours may be due to differences in staining techniques and methods of assessment; unlike other studies we did not quantify the number of affected fibres. It has been suggested that a further biopsy be considered to confirm the diagnosis if the Griggs criteria are not satisfied at first sight.5 ,11 We found that this is unlikely to achieve a diagnosis in the majority of these patients and is unnecessary when the characteristic pattern of weakness is present and the histopathological findings supportive of, and not inconsistent with, the diagnosis of IBM.
The Griggs criteria were widely considered to be the gold standard diagnostic tool for IBM. They have been instrumental in helping to identify the clinical characteristics particular to IBM. However, they lack sensitivity; 73% of our patients failed to fulfil them on initial assessment. Subsequently, the 1997 ENMC criteria were published.6 These place less emphasis on the histopathological features and therefore are more sensitive.27 Although these criteria were an improvement on the Griggs criteria they do not incorporate the characteristic lower limb pattern of weakness, which we found to be more common than finger flexor weakness at presentation, and they still require the presence of some of Griggs’ histopathological features. The advantage of the 2011 ENMC criteria is that they are more sensitive than the Griggs criteria and the 1997 ENMC criteria; the addition of the lower limb pattern of weakness, limits on age and CK makes them more specific. Therefore, the diagnosis of IBM can be made securely, even in the absence of Griggs’ histopathological features. Although the majority of patients fulfilling the Griggs criteria also have the diagnostic pattern of weakness associated with IBM, inevitably some patients with IBM will not satisfy the clinical criteria. A further strength of the 2011 ENMC criteria is that a diagnosis of clinicopathologically defined IBM can be made when the diagnostic pathological features are present in combination with supportive clinical features. This complementary approach means that the 2011 ENMC criteria are the most specific and sensitive criteria to date. However, some may feel that predominantly clinical criteria lack specificity and may lead to the inclusion of patients without IBM in studies. In our experience, the characteristic pattern of weakness associated with IBM is not seen in conditions important in the differential diagnosis of IBM. To confirm this, we reviewed the clinical examination findings, at presentation and with follow-up, in myopathies considered important in the histopathological differential diagnosis of IBM, including 15 cases of steroid responsive myositis, two cases of hereditary inclusion body myopathy and 10 cases of myofibrillar myopathy. None of these patients exhibited the characteristic pattern included in the 2011 ENMC criteria, suggesting that these clinical criteria do have sufficient specificity to differentiate IBM from its mimics.
We acknowledge the limitations to our study. It could be argued that to confirm the sensitivity of the clinical criteria a further biopsy should have been performed in those patients that did not fulfil the pathological criteria at presentation. However, we have shown that clinically diagnosed patients are indistinguishable from those with Griggs histopathological features and therefore repeating the muscle biopsy is unnecessary. Currently, in the absence of a ‘gold standard’ diagnostic test for IBM, we feel that clinical assessment with a supportive biopsy should be considered the ‘gold standard’ rather than the histopathological findings present in a small biopsy sample. Patients in our region with neuromuscular disease are referred early to specialist neuromuscular services which are not universally available. The majority of patients were assessed and managed by a single individual, possibly affecting the speed of diagnosis and management. Having a single assessor also had the advantage of uniformity of the clinical assessment and data collection. The study was retrospective which could introduce patient recall bias, particularly with regard to timings in this slowly progressive disease. Because many patients were followed-up, a complete dataset was collected for nearly every patient. In addition, we included all patients with a final diagnosis of IBM, not only those presenting with typical IBM. As the majority of our patients did not receive immunosuppressive therapy the results from our study are an accurate representation of the natural history of IBM.
Diagnostic criteria are useful in defining homogeneous cohorts for research purposes but are often impractical for everyday clinical use. We have shown that the characteristic pattern of weakness associated with IBM is found in the majority of patients at presentation and that this is independent of the histopathological features, supporting the diagnostic utility of the clinical examination and the addition of a clinically defined category to the diagnostic criteria. In addition, continued adherence to histopathologically predominant criteria will lead to misdiagnosis and inappropriate treatment of many patients with IBM and exclude a substantial proportion of patients from research studies, potentially leading to erroneous conclusions being drawn about the pathogenesis and treatment outcomes in IBM. Our data reveal that the clinical examination is highly sensitive and specific for IBM and that the majority of patients will not show all of the histopathological features previously considered to be diagnostic of IBM. However, we believe that the clinical and pathological assessments are complementary to one another and we continue to include muscle biopsy as part of the diagnostic assessment. The majority of patients will show some features consistent with the diagnosis on muscle biopsy and these are included in the proposed clinical diagnostic criteria. Whether the inclusion of newer immunohistochemical techniques such as p62 staining will lead to a further improvement in the histopathological diagnosis is currently uncertain. Very rarely, biopsy may reveal an unexpected alternative diagnosis. We are aware of patients who have been suspected by non-muscle specialists as having IBM but biopsy has led to alternative diagnoses, including myofibrillar myopathy, with a specific mutation identified, or granulomatous myopathy (sarcoid). This supports the continued inclusion of muscle biopsy in the diagnostic workup, especially if there is any clinical uncertainty.
Acknowledgments
SB is supported by the Muscular Dystrophy Campaign.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table
Footnotes
-
Correction notice This paper has been amended since it was published Online First. The authors have noticed a small error in one of the tables. In table 1, ’Proposed 2011 European Neuromuscular Centre diagnostic criteria for inclusion body myositis’, in the third column, ‘Upregulation of MHC class I’ has been removed.
-
Contributors SB: study concept and design, acquisition of the data, analysis and interpretation of the data, and drafting of the manuscript. WS: critical revision of the manuscript for important intellectual content and study supervision. DH-J: study concept and design, critical revision of the manuscript for important intellectual content and study supervision.
-
Competing interests None.
-
Ethics approval The study was approved by the Oxford University Hospitals NHS Trust Regional Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.