Article Text

Statistics from Altmetric.com
Comatose patients present to the neurologist in several ways, particularly in the emergency room or intensive care unit. Given the many causes of coma, the central focus is a comprehensive evaluation that starts with a detailed history, although this has to be an account—at least to begin with—from bystanders, paramedics or even the police. This is followed by a neurological and general examination, gathering of key findings, localisation of the involved brain structure, neuroimaging and other laboratory tests. Sorting out the cause of coma is what neurologists do best— compared with other specialists. Of course the relatively recent availability of MRI has helped tremendously in understanding some cases of coma but brain imaging can be normal. Indeed, unravelling the cause of coma remains in many circumstances a clinical judgement.
Once treated, comatose patients can recover suddenly (eg, after quick correction of hypoglycaemia) but many patients have significant structural brain injury and take days to awaken. About 25% of patients stay in a prolonged state of unconsciousness but even then slow awakening is to be expected; only a very small fraction survive but never emerge from coma.
Definitions of Altered States of Consciousness
Impaired consciousness has been traditionally regarded as a problem with alertness, awareness of self, or both.
A disturbance of arousal—mainly a function of the reticular formation in the brainstem— leads to diminished alertness.
A disturbance involving content—a function of multiple cortical areas—leads to diminished awareness, and failure to accurately integrate what is perceived.
These two components are interrelated but sometimes dissociated. One can be awake and aware, awake but not aware and not awake and not aware.
Coma is best defined as a completely unaware patient unresponsive to external stimuli with only eye opening to pain with no eye tracking or fixation, and limb withdrawal to a noxious stimulus at best (often with reflex motor movements). Brainstem reflexes can be intact or absent.
The degree of coma is more difficult to define. Terms such as stupor, drowsiness, obtundation, somnolence, light or deep coma are all vague and should be discouraged. It is better to note a set of key findings and make comparisons over time (eg, “the patient has improved, is now able to track my finger, but eyes open only to a loud voice, localise to pain, extubated and able to protect the airway”).
Comatose patients have several outcomes: they may lose all brain function (brain death), remain unconscious (persistent vegetative state) or regain consciousness (minimally conscious, fully conscious but disabled, or good recovery) (figure 1). Most emerge from coma in 1–2 weeks. Survivors with abnormal consciousness may remain in a minimally conscious state (MCS) or in a persistent vegetative state (PVS). Empirically, there is overlap between these states, particularly in the first weeks to months after the acute brain injury. Some patients may have only a fraction of awareness—for example, brief fixation to objects that is only detected by close observation over time. These conditions are not diagnosed on the spot.

Categories of altered states of consciousness, from coma to its potential consequences. MCS, minimally conscious state; PVS, persistent vegetative state.
Persistent vegetative state (PVS) is defined as:
No awareness of self or environment.
No sustained reproducible, purposeful or voluntary behavioural response to visual, auditory, tactile or noxious stimuli.
No language comprehension or expression.
Mostly intact cranial nerve reflexes.
Roving nystagmoid eye movements.
Presence of sleep and wake cycles, often eyes open during the day.
Stable unsupported blood pressure and intact respiratory drive.
Bowel and bladder incontinence.
Minimally conscious state (MCS) is defined as:
Patients make eye contact or turn head when being talked to.
An abulic emotionless state but with eye tracking movements.
May mouth words, may fend off pain.
Eyes following moving person.
Some intelligible verbalisation.
May hold object or use object when asked.
The frequency of these prolonged comatose states is not known, nor is it clearly established where the point of no return is. Patient registries may provide some insight into the frequency of coma after severe traumatic brain injury or stroke but there are definitional difficulties and confounding factors; for example, sedation in the first weeks after major brain injury (physician induced coma) may impact on time to awakening. Here are some sobering facts.
Withdrawal of support is a common cause of death in comatose patients.
MCS is far more common than PVS within 6 months from brain injury.
The diagnosis of PVS may not have been made by a neurologist—misjudgements are more common than appreciated.
Patients in PVS may be kept alive for decades but with no prospects for improvement.
Anatomy and Neurophysiology of Coma
Coma is currently understood as being caused by interruption of the ascending reticular activating system in the midbrain and pons projecting to the thalamus and cortex.
The thalamus through its intralaminar nuclei plays an important role in maintaining arousal.
The thalamus and ascending reticular activating system can be damaged from shift or displacement of the brainstem, as well as by direct destruction.
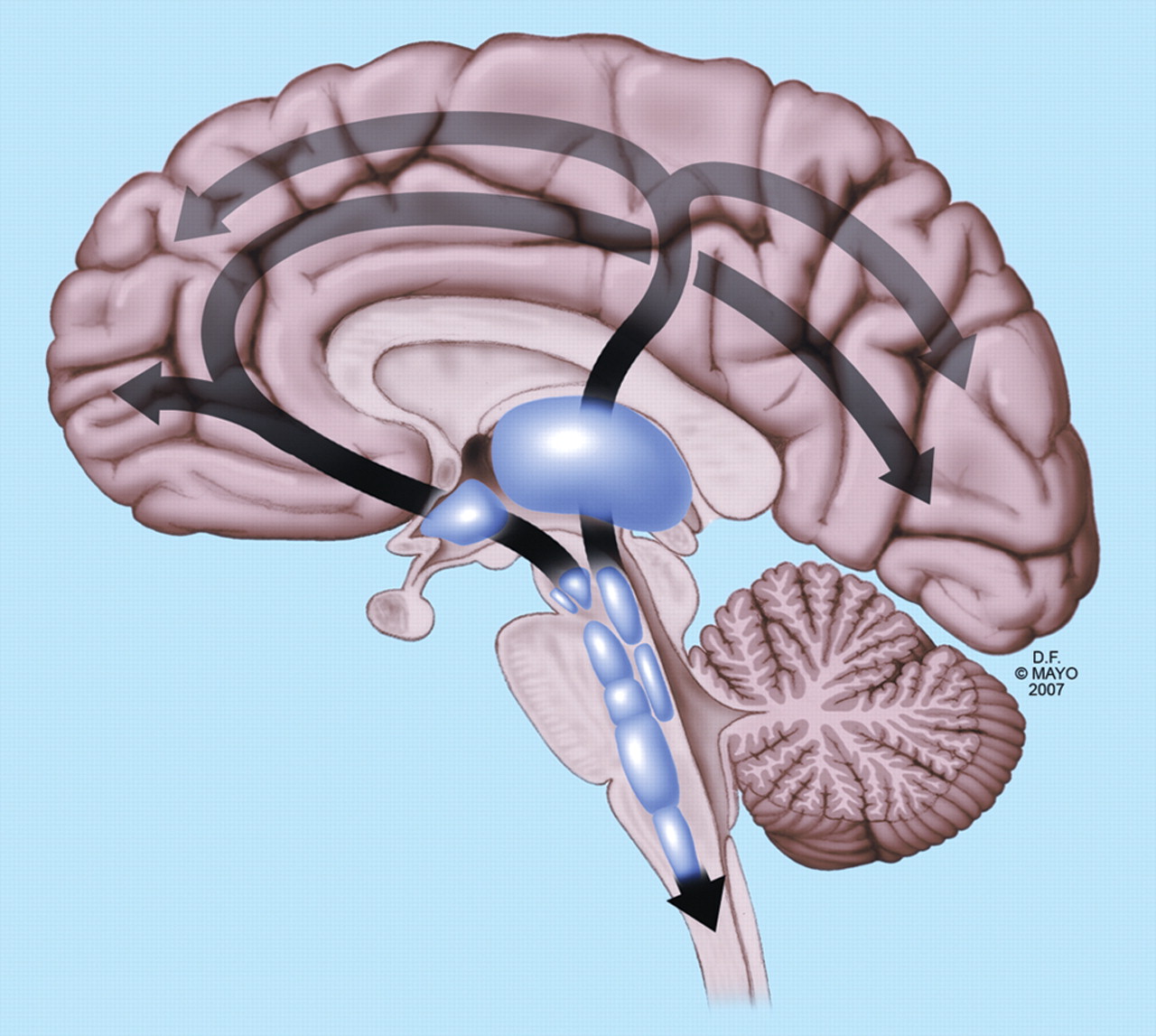
The ascending reticular activating system contains cholinergic neurons of the medial pontine tegmentum projecting to the thalamus, and monoaminergic neurons projecting from the upper brainstem to the thalamus, hypothalamus, basal forebrain and cortex (figure 2).
Stimulation of the posterior hypothalamus causes arousal.
Lesions of the cuneus and precuneus association cortex may be involved in the MCS.
Lesions of the anterior cingulate gyrus result in marked abulia.

The main pathways connecting the ascending reticular formation with the thalamus and cortex. From The comatose patient, Wijdicks EFM, used with permission of Mayo Foundation for medical education and research.
Neurological Examination of the Comatose Patient
Finding the cause of coma requires many steps and it helps to know the major categories before starting (table 1).
Classification and causes of coma
Structural injury of the cerebral hemisphere(s).
Intrinsic brainstem injury, or compression from surrounding damaged tissue such as swollen infarcted cerebellum.
Acute metabolic or endocrine derangement (eg, hypoglycaemia, hyponatraemia, acute panhypopituitarism).
Diffuse physiological brain dysfunction (eg, seizures, intoxication or poisoning, hypothermia, smoke inhalation, near drowning, heat stroke, acute catatonia, malignant neuroleptic syndrome).
Every physician should be familiar with two pitfalls.
The first is failure to recognise the locked-in syndrome which can mimic coma. (Some patients in this state have been inadvertently insufficiently sedated, intubated and pharmaceutically paralysed.) In a classic form of locked-in syndrome, they have their eyes open and can only blink to commands or move their eyes vertically. Their motor tracts are deefferented by a lesion in the ventral pons sparing the ascending reticular activating system and thus they can hear, see and feel pain.
The second, psychogenic unresponsiveness, is often only considered after exclusion of other causes and requires some clinical experience to definitively demonstrate its presence and to think about it in the first place and before ordering a battery of tests, some of which may be risky. The hand drop test is a classic but not completely reliable test (one arm is lifted and held in front of the face and when let loose slides next to the patient’s face rather than on to it). Closed eyes which open with tickling the nose hairs is more characteristic. Some patients may have forced upward or downward gaze that may suddenly change in direction. Others have abnormal movements, misinterpreted as seizures, but these generally look more like ‘fish out of water’ flopping. (To complicate matters, I have seen a psychogenic locked-in syndrome with blinking as the only form of communication in a nurse assistant for a full 48 h after a marital spat.)
Examination of the comatose patient follows a set of clinical tests to locate the lesion. These findings are then integrated with neuroimaging or electrophysiology test results. Many comatose patients do not, however, have specific findings which can be problematic. Nonetheless, a routine set of clinical tests is necessary to assess the depth of coma, the location of the lesion and possibly the underlying cause.
Initial neurological examination may use a coma scale—the Glasgow Coma Scale is a universally used instrument (table 2).
Another coma scale, the FOUR score, provides far more neurological detail while incorporating the bare necessities of a coma examination: eye response, motor response, brainstem reflexes and respiration (table 3).
Glasgow Coma Scale
The FOUR Score
The neurological examination proceeds with assessment of the cranial nerves and motor response to pain.
Fundoscopy may reveal diagnostic findings (eg, subhyloid haemorrhage in aneurysmal subarachnoid haemorrhage, acute papilloedema in increased intracranial pressure or hypertensive crisis).
Small or pinhole pupils (<2 mm) are due to a pontine lesion or opioids (figure 3A).
Midsize light fixed pupils (4–6 mm) are due to a midbrain lesion (figure 3B).
Maximally dilated pupils (>8 mm) are due to a lesion of the third cranial nerve nuclei, mesencephalon or compression of peripheral fibres of the third nerve. However, drugs and toxins can also dilate the pupils (eg, lidocaine, amphetamines, cocaine).
Unilateral fixed pupil is due to a third cranial nerve lesion (compression of the midbrain ocular motor complex, traction on the third nerve or pressure of the nerve against the clivus).
Spontaneous eye movement abnormalities (ping-pong, ocular dipping) localise mostly to bihemispheric dysfunction except ocular bobbing (rapid down, slow up) which localises to the pons.
Roving eye movements indicate that the brainstem is intact.
Skew deviation of the eyes suggests an acute brainstem injury.
Horizontal deviation of the eyes to one side might be a sign of non-convulsive status epilepticus but also of an ipsilateral hemispheric or contralateral pontine stroke.

(A) Pinpoint pupils: well known to any inner city police officer (opioid intoxication) but may also indicate pontine haemorrhage. (B) Mid position light fixed pupils (mesencephalic lesion) in downward compression of the upper brainstem from a hemispheric mass but also often the first sign of loss of all brainstem reflexes (brain death).
Evaluation of breathing pattern abnormalities may be useful.
Cheyne–Stokes breathing can occur with any type of reduced alertness and is least localising.
Cluster breathing and central neurogenic hyperventilation indicate bihemisphere or pontine lesions, while ataxic breathing indicates a lesion in the lateral tegmentum of the lower pons.
However, recognition of these patterns is difficult and cannot be observed when mechanical ventilation has taken over. Moreover, their localisation is not firmly established.
Motor responses are elicited with painful stimuli such as compression over the supraorbital nerve, temporomandibular joint or nailbed; flexion, extension or no response at all. None of these responses are localising and even the distinction between decerebrate and decorticate responses may not have significance for prognostication (not infrequently both responses can be present in the same patient).
Decorticate responses are defined by slow flexion of the elbow, wrist and fingers.
Decerebrate responses are defined by adduction and internal rotation of the shoulder, arm extension and wrist pronation with fist formation.
Spontaneous generalised myoclonus may indicate anoxic–ischaemic brain injury but also lithium intoxication, cephalosporin intoxication and pesticides.
Clues from the General Physical Examination
Although none is very specific and they can be seen in other circumstances, signs which suggest certain intoxications and metabolic abnormalities include the following:
Classic foul breaths are dirty toilet (uraemia), fruity sweat (ketoacidosis), musty or fishy (acute hepatic failure), onion (paraldehyde) and garlic (organophosphates).
Skin bullae may indicate barbiturate poisoning.
Dryness of skin indicates barbiturate poisoning or anticholinergic agents.
Profuse sweating indicates cholinergic poisoning, neuroleptic malignant syndrome or a serotonin syndrome.
Hypotension may indicate deliberate overdose with antihypertensives.
Hypertension may indicate amphetamine intoxication, cocaine or more or less any party drug.
Hypothermia (<35°C) is common in alcohol intoxication, and overdose with barbiturates or tricyclic antidepressants.
Hyperthermia (>40°C) can be seen with cocaine, tricyclic antidepressants, phencyclidine and salicylate intoxication.
Cardiac arrhythmias are a common manifestation of tricyclic antidepressant, cocaine and ethylene glycol poisoning.
The vital signs may also provide clinical clues to the diagnosis:
Hypothermia (<35°C) is commonly due to exposure to a cold environment but may also indicate decreased heat production due to hypothyroidism, Addison’s disease or hypoglycaemia. Hypothermia in comatose patients may herald brain death.
Hyperthermia (>40°C) indicates a fulminant systemic infection, endocarditis or CNS infection.
Hypotension may indicate early sepsis or fulminant acute meningococcal meningitis.
Hypertension may indicate that coma is due to acute hypertensive encephalopathy; it is also part of the Cushing reflex which is common in catastrophic intracerebral haemorrhage, in particular in the pons or cerebellum.
Finding the Cause of Coma
After the patient is medically stable, more inquiries should follow. Remember some questions may be inappropriate, others could be essential. Questions that might be asked of family members, paramedics, or bystanders include the following.
Could this be a major anoxic–ischaemic insult to the brain? How was the patient found? What did the scene look like when he or she was found? Was the patient breathing on arrival of the response team? Was a cardiac arrest documented? How long did the efforts last before resumption of the circulation? Was there noticeable blood loss? Did the patient deteriorate during transport?
Could this be an intoxication? What pills or over-the-counter drugs or herbs does the patient have access to? Has the patient made prior suicide attempts, has there been a psychiatric consultation, are there problems at work? Has anyone complained about drug or drinking habits?
Could this be a CNS infection? Did the patient use antibiotics for infection? Was there a rapid onset of fever and headache?
Could this be hypoglycaemia or hyperglycaemia? Is the patient known to be diabetic, or could they have undiagnosed diabetes?Has the patient had prior episodes of diabetic ketoacidosis, and has there been a recent change in diabetic medications? Might the patient have overdosed on hypoglycaemic drugs (their own or someone else’s)? Could they have been deliberately poisoned?
Might the patient be hyponatraemic? Did they have access to fluids and is he or she on diuretics?
Could this be non-convulsive status epilepticus? Is the patient a known epileptic?
Could this be an embolus to the basilar artery? Is the patient known to have atrial fibrillation or other cardiac disease and has warfarin recently been discontinued? Was hypertension poorly controlled?
The most important questions that need to be asked next are as follows.
Is the coma due to a destructive structural brain lesion or to global acute physiological derangement of brain function?
Is there any structural lesion in both cerebral hemispheres or in the brainstem?
Is there any brainstem lesion inside the brainstem or due to displacement of the brainstem? These syndromes may overlap:
Intrinsic brainstem lesions are recognised by skew deviation, internuclear ophthalmoplegia, small or unequal pupils and absent oculocephalic responses;
Brainstem displacement caused by lesions above the tentorium is recognised by a wide, fixed pupil, abnormal motor responses but otherwise intact brainstem reflexes;
Brainstem displacement from below the tentorium is recognised by small pupils, absent corneal reflexes and oculocephalic responses in some patients.
Patients with an intrinsic brainstem syndrome and a normal CT scan most likely have an embolus to the basilar artery; an abnormal CT scan will likely show a haemorrhage into the brainstem, traumatic brain injury to the brainstem or a brainstem tumour or infectious mass.
Patients with a brainstem displacement syndrome and an abnormal CT scan often have a massive hemispheric ischaemic or haemorrhagic stroke, contusional lesion, large subdural or epidural haematoma, a malignant tumour or abscess with oedema.
Is any bihemispheric syndrome due to cortical injury, extensive white matter injury or acute physiological brain dysfunction? Patients with bihemispheric lesions have few localising findings. Gaze preference may be seen temporarily. Brainstem reflexes are usually intact.
Patients with bihemispheric injury and a normal CT scan most likely are comatose from a toxin, status epilepticus, CNS infection, endocrine/metabolic derangement or anoxic–ischaemic injury.
In patients with a bihemispheric syndrome, an abnormal CT may show cortical lesions, white matter lesions, acute hydrocephalus, brain oedema, cerebral haemorrhage or posterior reversible encephalopathy syndrome.
Myoclonic status epilepticus may due to hypoxic–ischaemic brain injury after cardiopulmonary resuscitation.
Brain death is recognised by absent eye opening to pain, no motor response to pain, and absent pupil, cornea and corneal responses. Ocular vestibular reflexes are absent and there is no evidence of spontaneous breathing.
Investigations
Acute metabolic derangements (table 4) should be considered, such as hypoglycaemia, hypercalcaemia, hyponatraemia or hypernatraemia, acute uraemia or acute liver failure; panhypopituitarism and myxoedema coma are rare.
Laboratory values compatible with coma in patients with acute metabolic and endocrine derangements*
Essential initial laboratory tests include haematocrit, full blood count, platelet count and blood smear for schistocytes; electrolytes; glucose; urea; creatinine; liver function; osmolality; arterial blood gases; and thyroid function.
If intoxication is suspected, urine and blood screening is needed. Drug screens are however limited in scope and may not detect the toxin.
Blood cultures in patients with fever and suspicion of CNS infection.
Recognise metabolic acidosis and any anion gap due to methanol, ethanol, paraldehyde or salicylates. Calculate anion gap from the serum electrolytes. Anion gap = [Na+] – [Cl−] – [HCO3−]. A gap of 11–13 mEq/l is normally present but may increase with toxins.
The osmol gap is calculated using the equation: 2 × Na + glucose/18 + blood urea nitrogen/2.8. The calculated osmolality is normally less than the measured osmolality but should be no more than 10 mosmol/l. Atypical alcohols such as methanol, ethylene glycol (antifreeze) and isopropyl glycol increase the osmol gap.
CT and MRI of the brain are exceedingly important in the workup of a comatose patient— without them the cause of coma may remain a wild guess (table 5).
Frequent abnormalities on brain imaging in coma
Imaging defines the existence of any mass, its remote effect and any oedema, and may hint at the cause—for example, brain tumour, subdural haematoma, abscess.
CT will also detect fresh intracranial haemorrhage and contusions.
CT in traumatic brain injury is only a snapshot; in some cases repeat CT will show further abnormalities (figure 4).
MRI may show cortical injury, laminar necrosis, white matter disease and white matter oedema; it may be normal early after cardiopulmonary resuscitation.
MRI/MR angiography is the preferred test in patients with suspected acute basilar artery occlusion but many centres may more easily obtain a CT angiogram or go directly to catheter angiogram for possible neuroradiological intervention.

Rapidly evolving lesions on CT, pointing up the common requirement for repeat CT after traumatic brain injury. (A) Right subdural haematoma and contusional haemorrhages. (B) New contralateral extra dural haematoma.
In patients with a normal CT scan and no explanation for coma, a lumbar puncture must be done to rule out infection. This should include:
opening pressure;
description of the CSF appearance (cloudy, yellow, bloody, etc);
analysis for protein, cells and glucose;
CSF culture;
india ink stain and cryptococcal antigen;
viral titres and PCRs in CSF.
EEG has lost some of its diagnostic value in the evaluation of coma but is able to document non-convulsive status epilepticus in otherwise unexplained cases. Diffuse slowing and even the time honoured triphasic waves may be seen in patients with a structural cause of coma. EEG can be helpful in early herpes simplex encephalitis but MRI is much more specific in documenting the extent of the tissue destruction.
Management of Coma in the First Hour
The initial management of a comatose patient is to correct abnormal vital signs and laboratory abnormalities.
Improve oxygenation (face mask with 10 l/min oxygen flow aiming at a pulse oximeter saturation of >95%).
Intubate if patient cannot protect the airway (ie, increased work of breathing, pooling secretions, gurgling sounds).
Intubate any comatose patient with irregular ineffective respiratory drive and poor oxygenation.
Intubate any comatose patient with major facial injury or consider emergency tracheostomy.
Correct hypotension by placing patient in the Trendelenburg position and add crystalloids (rapid infusion of 500–1000 ml normal saline followed by 150 ml) and if no response, start vasopressors (phenylephrine intravenous bolus 100 μg/min).
Correct extreme hypertension (systolic above 250 mm Hg or mean arterial pressure above 130 mm Hg) with intravenous labetalol (10 mg IV), hydralazine (10 mg IV) or nicardipine (5 mg/h).
Correct hypothermia with warming blankets. However, consider induced hypothermia (33–34°C) treatment in patients who have been successfully resuscitated from cardiac arrest.
Correct hyperthermia with cooling blankets, icepacks and ice water lavage.
No harm is done if a patient with a high likelihood of hypoglycaemia is immediately given 50 ml of 50% glucose, even before the blood sugar is known (with coadministration of 100 mg thiamine intravenously).
No harm is done administering naloxone (0.4–2 mg every 3 min intravenously) if opioid intoxication is suspected.
No harm is done administering flumazenil (slow intravenous administration at 0.2 mg/min up to 5 mg) which effectively reverses any benzodiazepine toxicity; this however is contraindicated in patients with a seizure disorder and in whom concomitant tricyclic antidepressant intoxication is suspected (may provoke seizures).
Treat severe hyponatraemia with hypertonic saline and furosemide (3% hypertonic saline, 0.5 mg/kg hourly) after placing central venous catheter.
Treat hypercalcaemia with saline rehydration infusion followed by parenteral bisphosphonate pamidronate.
Consider elimination of any toxin by haemodialysis or haemoperfusion.
The priorities when faced with a patient with an acute structural cause of coma are as follows:
If any hydrocephalus, ventriculostomy to reduce raised intracranial pressure.
Consider evacuation of any mass to relieve increased intracranial pressure.
If a large mass cannot be removed, consider decompressive craniectomy; swollen brain will move out and decompress the mass effect on the deeper diencephalic structures.
Attempt to reduce suspected increased intracranial pressure with osmotic agents; mannitol, initial dose 1–2 g/kg intravenously, often with two repeated doses 30–40 min apart.
Then:
If CNS infection is even a remote possibility, start full coverage with cephotaxime (8–12 g daily, divided doses every 6 h intravenously), vancomyocin (2 g daily, divided dose every 12 h intravenously) and ampicillin (12 g daily, divided dose every 4 h intravenously) in combination with antiviral coverage with intravenous aciclovir (10 mg/kg every 8 h). Strongly consider intravenous dexamethasone 0.6 mg/ kg daily before administration of antibiotics, and continue for 4 days.
Long Term Management of Coma
If necessary, tracheostomy, percutaneous gastrostomy, bladder and bowel care, infection surveillance and deep venous thrombosis prophylaxis.
Patients may be weaned from the ventilator and tracheostomy removed later if secretions are controlled.
Methicillin resistant Staphylococcus aureus and vancomycin resistant Enterococci can be reduced by less overcrowding, strict isolation and avoidance of multiple antibiotics along with meticulous hand hygiene by staff.
Contractures can be reduced but not prevented by physical therapy.
Decubitus skin breaks and ulcers can be prevented by special beds and monitoring of pressure sites along with superb nursing care.
Neurostimulation is of no use in PVS and (as yet) unproven in MCS.
Dopaminergic agents (eg, bromocriptine), zolpidem and lamotrigine are commonly used but unproven stimulant drugs in patients with MCSs.
Outcome Prediction of Comatose Survivors
The outcome is determined by the underlying cause of coma. Outcome can also be subject to physician expectations; for example, if the situation is judged by a pessimistic neurologist as hopeless and care is withdrawn leading to death, this can be perceived as a self-fulfilling prophesy. Withdrawal of support or no aggressive care of complications in sick elderly patients may be more common than in prior healthy young patients. These clinical decisions may impact on the utility of prognostic models.
There are five prognostic indicators for poor outcome after anoxic–ischaemic brain injury:
myoclonic status epilepticus at any time;
absent corneal or pupil reflexes at 3 days;
absent motor responses at 3 days;
absent sensory evoked potential cortical responses;
increased serum neuron specific enolase at any time.
Outcome after traumatic brain injury is very difficult to predict. Many young patients make a good recovery despite severe CT scan abnormalities and slow progress.
Outcome after aneurysmal intracranial haemorrhage is determined by initial clinical grade; 50% recover to a better grade and some patients may fully recover.
Surgery is no better than medical management in deep ganglionic haemorrhage but evacuation of cerebellar haematoma can result in dramatic improvement. Deteriorating patients with an expanding lobar haematoma may benefit from evacuation but the degree of benefit is uncertain.
Outcome in CNS infections is affected by time to administration of antibiotic or antiviral drugs (and corticosteroids).
MCS may seem a better outcome for the family than PVS but may be a worse outcome for the patient who is then aware of the devastating injury.
Patients in MCS may further recover but no predictors are known.
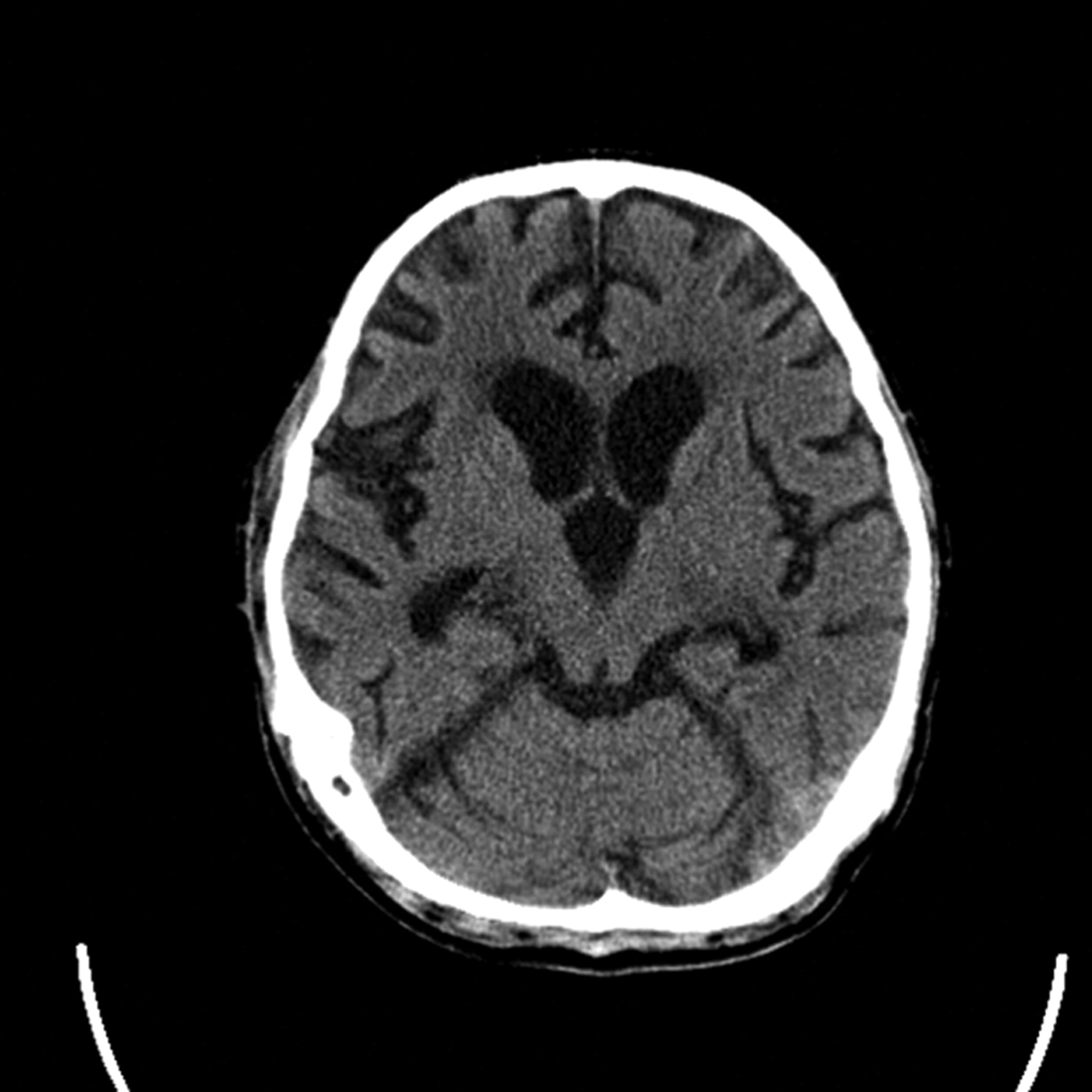
Patients in PVS for less than 3 years may rarely partially recover, and when they do it is mostly after traumatic brain injury. Nonetheless, all patients remain severely disabled. Patients in PVS for 3 years or more do not recover; they have remarkable generalised brain atrophy on CT scan (figure 5).

CT brain scan showing marked atrophy in a 26-year-old in persistent vegetative state after refractory status epilepticus (the CT appearance corresponds to the brain of a 100-year- old person).
Brain Death Determination
Brain death is declared when brainstem reflexes, motor responses and respiratory drive are absent in a normothermic, non drugged comatose patient with an irreversible widespread brain lesion of known cause and no contributing metabolic derangements.
In adults, brain death is a clinical diagnosis and confirmatory tests are not needed in the USA, UK and many other EU countries. One neurological examination should suffice but some hospital protocols may require two independent examinations and a certain period of observation. In children, confirmatory tests are required, and a longer time of observation.
Brain death means the patient has died and organ donation is allowed after consent of family members.
Prerequisites for brain death diagnosis (figure 6) include the following.
Identification of cause and irreversible coma, absent major confounding reversible medical illness and no lingering effect of pharmaceutical agents such as sedative drugs or neuromuscular blocking agents.
Core temperature ≥36.5°C, systolic blood pressure >90 mm Hg.
Euvolaemia, eucapnia, normoxaemia, normotension.
Absent brainstem reflexes.
Apnoea (no trigger of ventilator).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Stepwise determination of brain death.
Apnoea needs confirmation. The apnoea test is performed using the oxygen diffusion method. The patient is preoxygenated with 100% O2 followed by disconnection of the ventilator while delivering 100% O2 6 l/min through a suction catheter close to the level of the carina (approximately 1 cm beyond the end of the endotracheal tube). The test is positive, meaning no breathing effort, when the arterial PCO2 is ≥60 mm Hg (8 kPa) or if there is an increase of 20 mm Hg (3 kPa) in PCO2 above a normal baseline value.
Conclusions
Sorting out the cause and degree of coma remains in many circumstances a clinical judgment.
Ask the following seven questions. Could this be a major anoxic–ischaemic insult to the brain? Could this be an intoxication? Could this be a CNS infection? Could this be a major metabolic derangement or endocrine crisis? Could this be nonconvulsive status epilepticus? Could this be an embolus to the basilar artery? Could it be psychogenic?
CT (and CTA) MRI of the brain are important and may provide the answer (but not always).
The initial management is to correct abnormal vital signs and laboratory abnormalities.
The first priority with an acute structural cause of coma is treatment of increased intracranial pressure.
The first priorities with a possible CNS infection are broad antibiotic and antiviral coverage and corticosteroids.
Tracheostomy, percutaneous gastrostomy, bladder and bowel care, infection surveillance and deep venous thrombosis prophylaxis are key components of longer term care.
Outcome prediction in acutely comatose patients is determined by the underlying cause and may also be influenced by physician expectation.
To ensure that the cessation of brain function is irreversible, physicians must determine the cause of coma, exclude mimicking medical conditions, and perform a standardised series of tests.
Acknowledgments
This article was reviewed by Robin Howard, London, UK, and some of the specialist neurology registrars in Edinburgh, UK.
Footnotes
-
Competing interests None
-
Provenance and peer review Not commissioned; externally peer reviewed.
Other content recommended for you
- Predicting the outcome of a comatose patient at the bedside
- THE PROGNOSIS OF MEDICAL COMA
- On the difficulty of neurosurgical end of life decisions
- Somatosensory evoked potentials aid prediction after hypoxic–ischaemic brain injury
- Persistent vegetative state and minimally conscious state: ethical, legal and practical dilemmas
- Is it better to be minimally conscious than vegetative?
- Thalamic proton magnetic resonance spectroscopy in vegetative state induced by traumatic brain injury
- Hypoxic-ischaemic brain injury
- Confirmation of brainstem death
- Does it matter that organ donors are not dead? Ethical and policy implications