Article Text

Statistics from Altmetric.com
A 68-year-old man developed “pins and needles” of his fingertips and toes in December 2005 along with cramp-like muscle pains in his forearms and calves. Over the next 6 months he noticed “coldness” of his hands and feet, his arms felt “sore” and his fingers were often “shaky” and felt “gritty”. His feet felt as though they were “covered in sand” and he became unsteady. By June 2006 he was frequently falling. He described “not knowing where” his feet were and his “knees giving way”. By November 2006 his mobility was restricted to using a wheelchair. He also had difficulty with manual dexterity, leading to problems with dressing and feeding. Over this period he had intermittent diarrhoea and constipation but his weight and appetite were stable. His past medical history is outlined in table 1 and included longstanding abdominal pain and alteration in bowel habit for which he had been extensively investigated.
In November 2006 his medications included: fluoxetine 20 mg once daily for depression, omeprazole 20 mg once daily, folic acid 5 mg once daily, Adcal D3 three tablets once daily, ferrous sulphate 200 mg three times a day, alfacalcidol 1 μg once daily, hyoscine butlylbromide 10 mg three times a day, and intramuscular vitamin B12 injections 1 mg every 3 month which he had been receiving for the past 10 years. There was no family history of neurological illness. He was an ex-smoker who had given up 36 years previously, and he consumed alcohol occasionally.
On examination in November 2006 he was thin, weighing only 50.2 kg (body mass index 17.5 kg/m2). There was finger clubbing but no generalised lymphadenopathy and the rest of the general examination was unremarkable. He had normal higher mental function, cranial nerves and speech, muscle bulk and tone, and full power in all muscle groups. He had no tendon jerks and his plantars were downgoing. He had diminished distal sensation to pinprick in the upper limbs (up to the elbow on the left and mid forearm on the right) and legs (to the top of the left thigh and right knee). Joint position sensation was absent to the wrists and ankles. Vibration sensation was absent to the wrists and diminished up to the sternum. There were no cerebellar limb signs. Romberg’s sign was positive and he had a wide based ataxic gait.
The results of investigations are summarised in table 2.
Clinical neurophysiology
May 2006: mild axonal polyneuropathy with polyphasia on electromyography (EMG) suggesting a polyradiculopathy but some motor units appeared myopathic.
November 2006: smaller sensory responses in the hands compared with May 2006, suggesting an inflammatory process affecting the dorsal root ganglia but both sural responses were normal which, in conjunction with severe gait impairment, suggested immobility was not due to sensory neuropathy/dorsal root gangliopathy. Motor nerve conduction was normal, including f waves. EMG revealed some polyphasia typical of chronic neurogenic change. EMG abnormalities in the face of normal peripheral nerve conduction was indicative of involvement at the level of the nerve roots or anterior horn cells, or possibly primary pathology in the spinal cord.
Neuroimaging
MR imaging revealed multiple small white matter lesions in both cerebral hemispheres on T2 and FLAIR with some patchy ill-defined high signal within the pons and upper thoracic cord likely to be affecting the dorsal columns (fig 1).

MR imaging revealed areas of signal change in the dorsal columns of the cervical and thoracic spinal cord (A, B) (arrowed)—T2 weighted. In addition, there were multiple white matter changes in both cerebral hemispheres and the pons (C, D) (arrowed)—FLAIR.
Clinical progress
The presumed diagnosis at this point was of a dorsal root ganglionopathy. The patient received a course of intravenous immunoglobulin (IVIg) therapy in November 2006 with no benefit. Over the next 6 months he developed painful dysaesthesiae in his hands and feet with minimal relief from gabapentin and carbamazepine. His ataxia progressed and he was ultimately unable to transfer independently and required assistance with all activities of daily living. He experienced nausea and vomiting and his weight steadily declined. He was transfused 2 units of red cell concentrate every 3 months to maintain a haemoglobin level of over 100 g/l. He became bed bound and developed a lower respiratory tract infection which failed to respond to antibiotics and he died in June 2007. A post mortem was performed.
DR KEVIN TALBOT’S DISCUSSION
The essence of this case is of a man in his sixties who, over 18 months, developed a progressive neurological problem from which he ultimately died. In the background is a 20 year history of very complicated gastrointestinal problems. Our task is to define his neurological syndrome, identify the gastroenterological problem, and decide whether or not they were related.
In December 2005 he presented with what I presume was an insidious onset of pins and needles in his fingers and toes, and by 6 months into the illness he had developed significant mobility problems and complained of “not knowing where” his feet were and that his knees were “giving way”. These two symptoms suggest proprioceptive loss, the anatomical substrate of which is either in the spinal cord or peripheral nerves. Prominent involvement of the upper limbs might favour the spinal cord rather than the peripheral nerves but both are possible, as indeed is a mixture of the two. He had problems with manual dexterity and intermittent diarrhoea and constipation although his weight and appetite were stable. However, his weight had been low for the preceding two decades. Bladder or visual symptoms are not mentioned and therefore I assume these were absent. There was no family history of neurological disease. We are not told his occupation. He is just at the tail end of the age group who would have been conscripted for National Service and so it is worth considering whether this man had been abroad, to areas where he might have acquired specific infections. He used to smoke many years ago and did not drink much alcohol.
To make sense of the complex gastrointestinal history, it helps to represent it as a timeline (fig 2). He had undergone a tonsillectomy in childhood, which was common in that period, and probably irrelevant. About 20 years prior to his neurological presentation he developed gastrointestinal problems, initially abdominal pain and altered bowel habit, and he underwent a rectal biopsy, demonstrating spirochaetosis. We shall return to this result, as we clearly can not ignore it in the light of his subsequent neurological syndrome. A contemporary barium study showed “jejunal diverticulosis”. Having not previously encountered jejunal diverticulosis, I sought the expert advice of a gastroenterologist in understanding the relevance of this finding to the case under discussion.
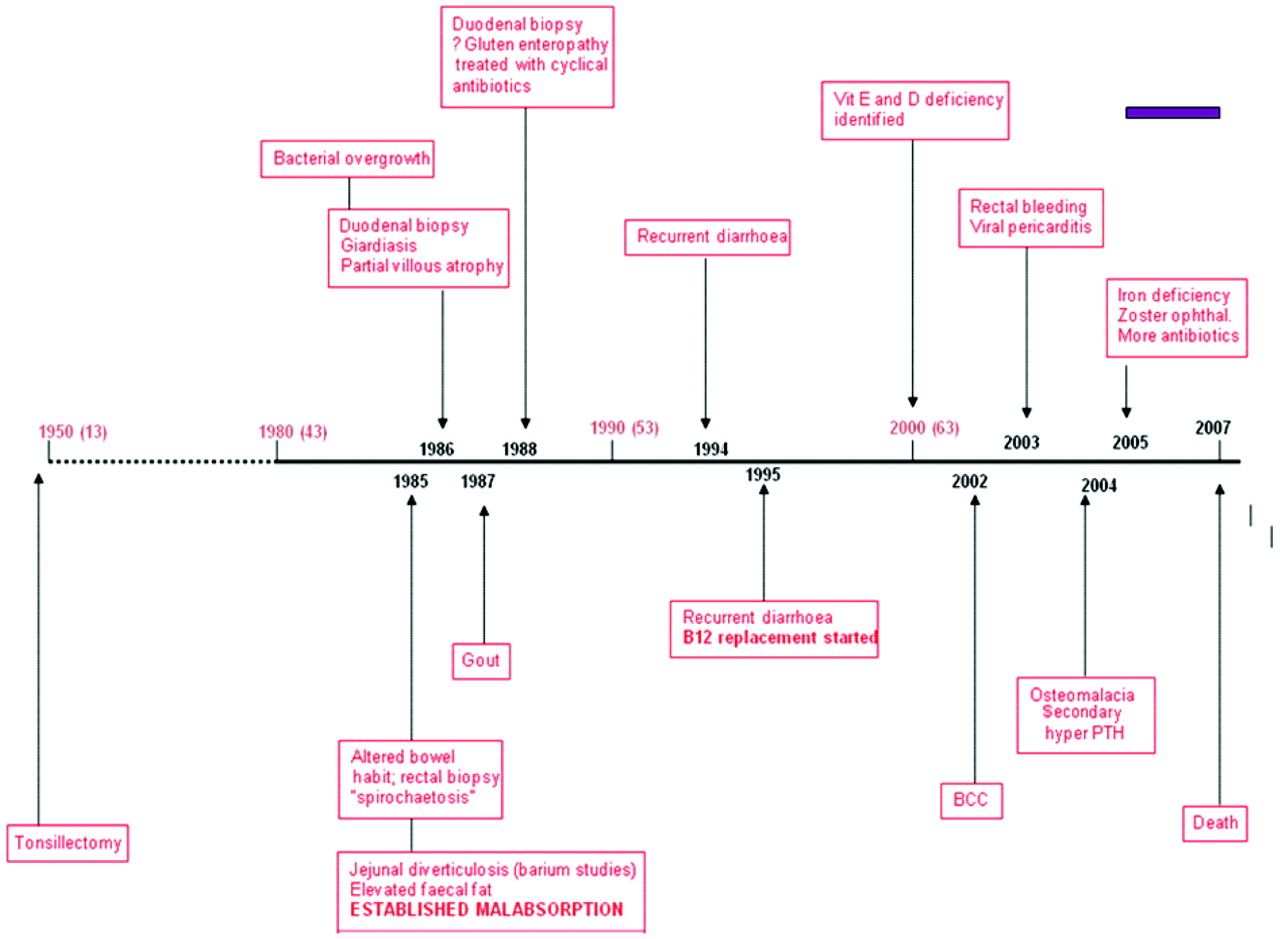
Timeline of gastrointestinal symptoms and other past medical history. BCC, basal cell carcinoma; PTH, parathyroidism.
The key fact is that by 1985 this man had established malabsorption with raised levels of faecal fat. A 20 year history of malabsorption is long enough to produce severe deficiency problems. Duodenal biopsies, in 1988 and 1996 for recurrent abdominal pain and diarrhoea, confirmed partial villous atrophy. Giardiasis was found in the first biopsy. Does this mean he did spend time abroad? Does it suggest immunodeficiency? Although it is difficult to believe it was not measured earlier, we are told that by 1995, 10 years after the clear documentation of malabsorption, his vitamin B12 was low. He was therefore started on replacement therapy a decade before the onset of his neurological problems. In 2000 he was found to be deficient in the fat soluble vitamins D and E. He developed viral pericarditis in 2003 but I cannot relate this to his other medical problems. Then, not unsurprisingly, he was shown to have osteomalacia and secondary hyperparathyroidism, a consequence of malabsorption. By 2005 he was iron deficient. He also developed zoster ophthalmicus; was this because he was generally debilitated or does it represent further evidence of an immune deficiency syndrome? Throughout the whole of the period of malabsorbtion he received recurrent courses of antibiotics, presumably for weeks or longer, most likely metronidazole and tetracyclines. His neurological decline and death (purple bar on fig 2) was a very short part of his overall 20 year history of malabsorption.
The examination findings described in November 2006 are well into the period of his neurological syndrome. Pseudoathetosis suggests dorsal column or peripheral nerve problems. The observation of normal muscle bulk, tone and power suggests that his disability was due to ongoing problems with proprioception rather than upper or lower motor neuron involvement. He was treated with IVIg in 2007, perhaps for presumed paraneoplastic syndrome or replacement for an underlying immune deficiency. Refractory bowel infections such as Clostridium difficile can respond well to IVIg. Despite this treatment, he continued to deteriorate neurologically as well as losing further weight. He also required blood transfusions to maintain his haemoglobin levels. In June 2007 he died of a chest infection, 18 months after the onset of his neurological symptoms.
In summary, this is the case of a 68-year-old man with progressive sensory ataxia on the background of a 20 year history of malabsorption. The underlying pathology appears to be either in the dorsal columns or the peripheral nerves, or both. I would favour the dorsal columns on the basis of the imaging.
The abnormal blood tests indicated recurrent anaemia with a fluctuating mean corpuscular volume. There are a number of different interpretations of this, including gastrointestinal bleeding and nutritional deficiency states, particularly of vitamin B12 and folate. He was consistently neutropenic and I think this is highly relevant. The blood film showed evidence of iron deficiency, burr cells and acanthocytes which occur in a range of conditions, including hyposplenism, coeliac disease and vitamin E deficiency (his levels were low). The remainder of the blood tests showed no evidence of liver or kidney problems and no evidence of a systemic inflammatory response, his immunoglobulin profile was normal and there were a number of negative antibody tests, including antineuronal antibodies (in particular anti-Hu). Duodenal biopsies showed partial villous atrophy.
The neuroimaging demonstrated high signal in the dorsal columns (seen on axial and sagittal sections) suggesting a focal process which can be interpreted in this context as Wallerian degeneration with gliosis. There was no evidence of cerebellar degeneration and the multiple white matter changes in both cerebral hemispheres and pons may have represented small vessel ischaemic changes.
The neurophysiology aimed to differentiate whether his sensory ataxia was due to a peripheral neuropathy or central pathology in the spinal cord. There was evidence of a mild axonal polyneuropathy and of dorsal root ganglia involvement but the salient feature is that these were mild, and certainly not enough to explain why this man could not walk, providing support for a process which was much more focused on the spinal cord.
Examination of the CSF was normal with nothing to suggest an infective or inflammatory process. We are not told about syphilis serology which, given the “spirochaetosis” on the rectal biopsy, would have been relevant. Neither did he have an HIV test as far as we have been informed. Although a normal CSF can be found in tabes dorsalis, it does make an active CNS infection or paraneoplastic disorder unlikely.
So the key questions are:
What is the cause of the malabsorption?
What are the consequences of the malabsorption for the CNS in terms of multiple deficiencies, including vitamins B12, E, A and D, and iron and others.
Malabsorption can arise as a result of various mucosal disorders, such as gluten sensitive enteropathy with total or partial villous atrophy. In fact, partial villous atrophy is non-specific and is not synonymous with coeliac disease but can also be secondary to a variety of other gastrointestinal pathologies. Additionally, crypt hyperplasia and lymphocytic infiltration are required on histology to confirm coeliac disease. The terminal ileum is a crucial site of absorption and can be affected in a number of conditions, including tuberculosis and Crohn’s disease. Malabsorption can also occur as a result of structural bowel problems (eg, following bowel resection) or in the presence of blind loops and diverticulae where the mechanism is bacterial overgrowth. There may also be very specific mechanisms for malabsorption (eg, autoimmune loss of intrinsic factor).
Was this gluten sensitive enteropathy? I don’t think so. Partial villous atrophy is non-specific and he had two negative antibody tests. To definitively establish a diagnosis of coeliac disease it is necessary to have a period of freedom from gluten followed by normalisation of the mucosal histology followed by rechallenge with a gluten diet and another biopsy with typical findings of gluten sensitivity. Although controversial, a number of neurological associations of gluten sensitivity have been reported, including ataxia, peripheral neuropathy and epilepsy but these are not features in this case.1 So I will discount coeliac disease on clinical grounds.
Tropical sprue is a post-infective malabsorption syndrome. Giardia is quite a common precipitant. It usually occurs in people who live in the tropics for a prolonged period of time. Complex bacterial infections may cause bacterial overgrowth of multiple organisms, and it can be extremely refractory to treatment. There may be extensive weight loss over a period of several years, as in this case. Without specific information that this patient had travelled abroad, I am unable to make the diagnosis of tropical sprue, but it may fit.
Whipple’s disease should be mentioned but the bowel pathology and neurological presentation in that condition are both very different to the current case.
Jejunal diverticulae occur along the mesenteric border of the jejunum and offer a better explanation for malabsorption in this case.2 Diverticulae are thought to be due to increased intraluminal “pulsion” pressure in people who have some underlying anatomical susceptibility. As a result, patients acquire diverticulae over the years which behave like multiple blind loops. This can result in an acute abdomen due to bleeding, infarction or infection, and could explain many of this patient’s symptoms, such as recurrent abdominal pain and altered bowel habit. The associated bacterial overgrowth has a specific toxic effect on enterocytes resulting in malabsorption, partial villous atrophy arising as a result of inflammation and even leads to consumption of nutrients by bacteria. So, I think the gastroenterological diagnosis here is acquired jejunal diverticulosis, chronic bacterial overgrowth and malabsorption.
Let us now consider some possible causes of the neurological syndrome.
Paraneoplastic syndromes have to be mentioned, if only to be dismissed. There was no evidence of a tumour although neither this nor negative antineuronal antibodies completely excludes the diagnosis. A condition called paraneoplastic myelopathy, which I have never encountered, is said to be an aggressive necrotising process and would not be confined to the dorsal columns as in this case. Overall I think there are better explanations for the neurological picture.
What about the possibility of neurosyphilis? This man had evidence of spirochaetes on rectal biopsy. Tabes dorsalis is very uncommon in the post antibiotic era but it has not completely gone away.3 It may follow a latent period of 15–35 years post-infection and the classic descriptions include “lightening pains” lasting from hours to days as well as gastric crises, autonomic involvement, areflexia, extensor plantars, Argyll Robertson pupils and optic atrophy. Although the phenotype of neurosyphilis may have changed in recent years and there are hints of tabetic features in this patient’s presentation, again I do not think it really fits clinically. Spirochaetes are apparently frequently found at rectal biopsy as an incidental finding.
Subacute combined degeneration of the cord involves the dorsal columns and corticospinal tracts. Patients initially present with paraesthesia in the hands followed by a proprioceptive ataxia and then a progressive spastic parapaparesis. The radiological findings here are strikingly similar to those described in the literature, with marked signal change in the dorsal cord. Cobalamin is the active molecule underlying vitamin B12 and it is a cofactor in DNA synthesis which affects the bone marrow resulting in anaemia. It also catalyses the isomerisation of methylmalonyl-CoA to succinyl-CoA. Serum vitamin B12 levels do not necessarily correlate with activity of B12 at a cellular level. In order to assess B12 functionality, it is necessary to measure levels of plasma homocysteine and methylmalonic acid which were not measured in this case. A raised methylmalonic acid level would indicate lack of conversion by vitamin B12 to succinate and hence functional B12 deficiency. The dorsal columns are affected more than the corticospinal tracts which in turn are affected more than the peripheral nerves. Haematological abnormalities, including a raised mean corpuscular volume, do not occur in all patients. Interestingly, vitamin B12 deprived monkeys do not develop anaemia or peripheral neuropathy but do develop dorsal column disease. However, I don’t think this was B12 deficiency because the patient’s B12 levels were consistently high and I have no prior expectation that he would have functional B12 deficiency. He had malabsorption and was treated with parenteral B12 for 10 years before developing neurological symptoms. So, although I think this is the clinical and radiological syndrome of subacute combined degeneration, it is not due to B12 deficiency
What could cause something similar? We know his vitamin E levels were low. The neurological manifestations of vitamin E deficiency are complex but include sensory ataxia. One of the conditions I considered was common variable immune deficiency, an acquired disorder characterised by pan-immunoglobulin deficiency resulting in recurrent infections and granulomatosis. It is associated with partial villous atrophy and neurological complications, including unsteady gait, and can cause a peripheral neuropathy, which we know was present in a mild form in this case. However, vitamin E deficiency causes more widespread neurological abnormalities and while it may have been a contributory factor, I do not think it explains the entire presentation, and in any event he was probably given replacement therapy.
So what is the real diagnosis? There is a disease which may be relevant here called enzootic ataxia, first described in ruminants in Australia the 1930s.4 It is also known as “swayback” because affected sheep and cows sway backwards and fall over. It has many of the same features as the case under discussion. The cervical cord is most affected and there is Wallerian degeneration with demyelination and microcavitation of the dorsal columns. It may be precipitated by zinc supplementation, or when grazing in pastures deficient in copper. I think the neurological diagnosis in this patient was copper deficiency myelopathy caused by chronic malabsorption which in turn was caused by bacterial overgrowth due to jejunal diverticulae. He could have received zinc supplementation which may have precipitated the neurological decline. Other contributing factors may have been coexisting vitamin E deficiency and metronidazole induced peripheral neuropathy. However, I think everything can be explained by copper deficiency myelopathy. This condition has been brought into prominence by Kumar et al at the Mayo Clinic.5, 6 The clinical picture is characterised by prominent sensory ataxia associated with mild spasticity and peripheral neuropathy which is a variable feature (ie, the same clinical syndrome as subacute combined degeneration of the spinal cord). Approximately half of the patients have dorsal column signal change on MRI.7 The haematological features differ from B12 deficiency in that there is anaemia with associated neutropenia with a left shift in granulocytic and erythroid maturation with vacuolated precursors and ringed sideroblasts in the bone marrow.5, 6
Malabsorption and gastrointestinal surgery are common precipitants, as is zinc supplementation, and the condition affects a wide age range. Serum copper is either undetectable or extremely low; serum caeuroplasmin is also low with normal urinary copper excretion. Patients may also be anaemic. The anaemia responds rapidly to copper supplementation but resolution of the neurological deficits is much more variable and the functional state of many of the patients remains static.5, 6
The real problem I have with diagnosing this condition is that, of the 30 or so patients reported in the literature, none seem to have died from it. Is this the first patient to die and the first autopsy to be reported? Or have I just got this completely wrong? I think the clinical features are typical, he has exactly the right predisposing factors and it explains his anaemia. I suspect he died because he was very debilitated. I am not sure if the diagnosis was achieved in life and even if it was, copper replacement may not have halted his neurological deterioration.
DISCUSSION
Dr Ravi Malhotra (Consultant Neurologist): I would like to thank the patient who was a very cheerful and stoic gentleman in life. He had a background in the parachute regiment of the army and true to their tradition he was a Spartan. His wife kindly agreed to a post-mortem examination. My clinical impression was of a dorsal root ganglionopathy, possibly secondary to paraneoplasia. I did not consider the diagnosis of copper deficiency myelopathy.
Dr Greg Moran (Consultant Clinical Neurophysiologist): I think if you see someone with fairly normal sensory responses in the feet but they are unable to walk, a primary dorsal root ganglionopathy is very unlikely.
Professor Charles Warlow (Professor of Medical Neurology): Could this possibly have been a paraneoplastic neuropathy?
Dr Jeremy Rees (Consultant Neuro-oncologist): The clinical progression was too slow for a paraneoplastic neuropathy. It took a year or so for progression from walking to a wheelchair. Paraneoplastic neuropathy is usually asymmetrical and rapidly progressing.
NEUROPATHOLOGY: DR COLIN SMITH
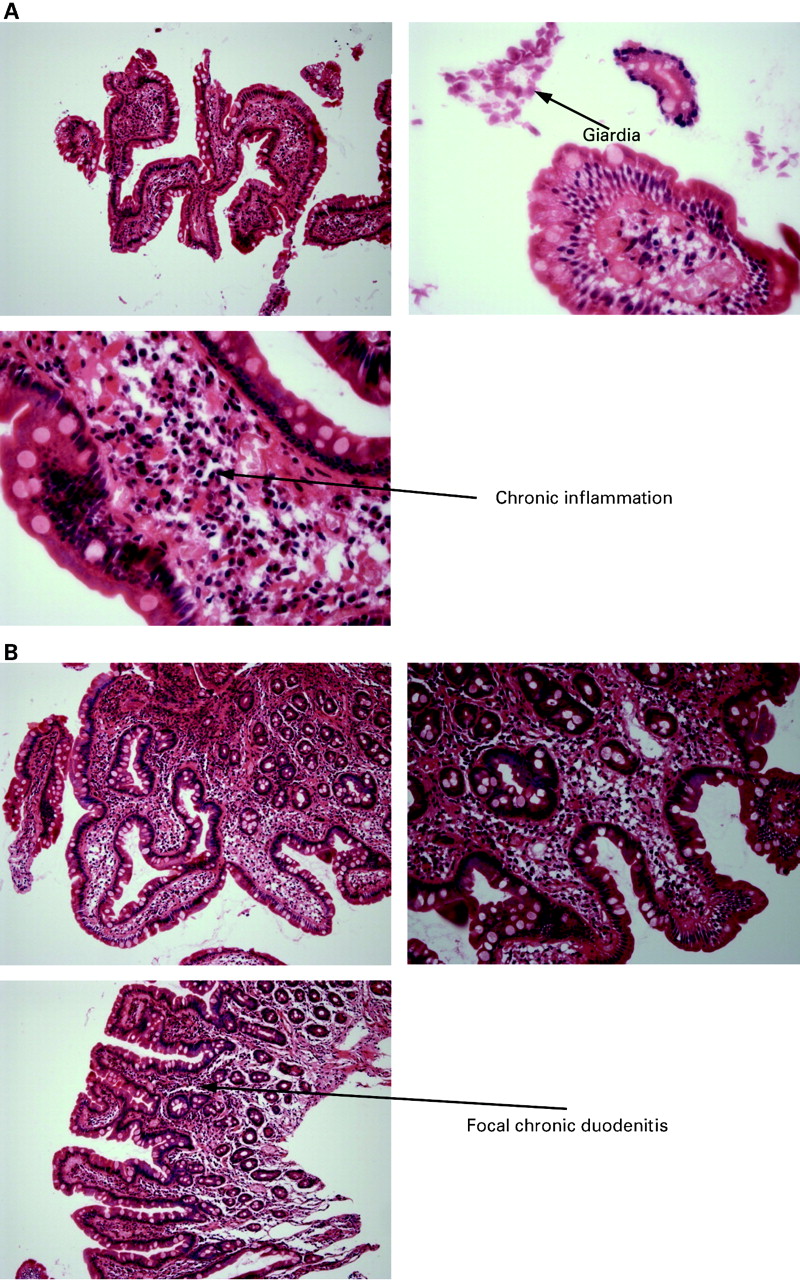
The duodenal biopsy of 1986 demonstrated partial villous atrophy with chronic inflammation in the lamina propria and giardiasis (fig 3A). A repeat biopsy from 1988 demonstrated mild partial villous atrophy, a non-specific term describing abnormalities of uncertain significance (fig 3B), possibly representing focal chronic duodenitis following elimination of giardia or incomplete resolution of a gluten sensitive enteropathy. There were no pathological features of coeliac disease and, in particular, there was no evidence of intraepithelial lymphocytes or crypt hyperplasia. Biopsies from 2005 included a normal terminal ileal biopsy, a colonic biopsy which demonstrated melanosis coli and a biopsy from the second part of the duodenum which was described as showing mild duodenitis but once again with no pathological features of coeliac disease. Therefore, the pathological diagnoses by 2005 included previous giardiasias and duodenal villous atrophy with focal chronic duodenitis but no pathological features of coeliac disease. The changes may represent non-specific inflammation or ongoing infection.
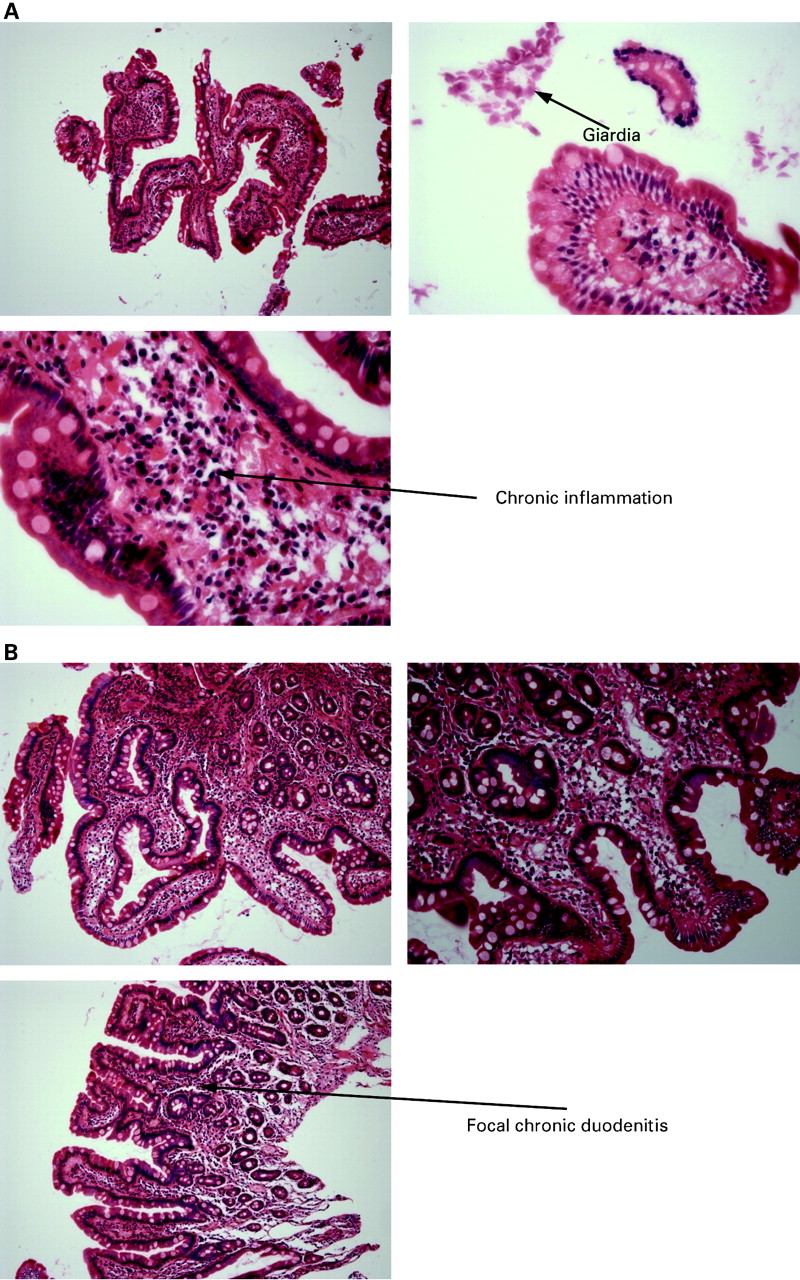
(A) Haematoxylin–eosin (H&E) stained photomicrographs from the duodenal biopsy showing chronic inflammation, partial villous atrophy and giardiasis. (B). H&E stain of the duodenal biopsy showing mild partial villous atrophy with focal chronic duodenitis.
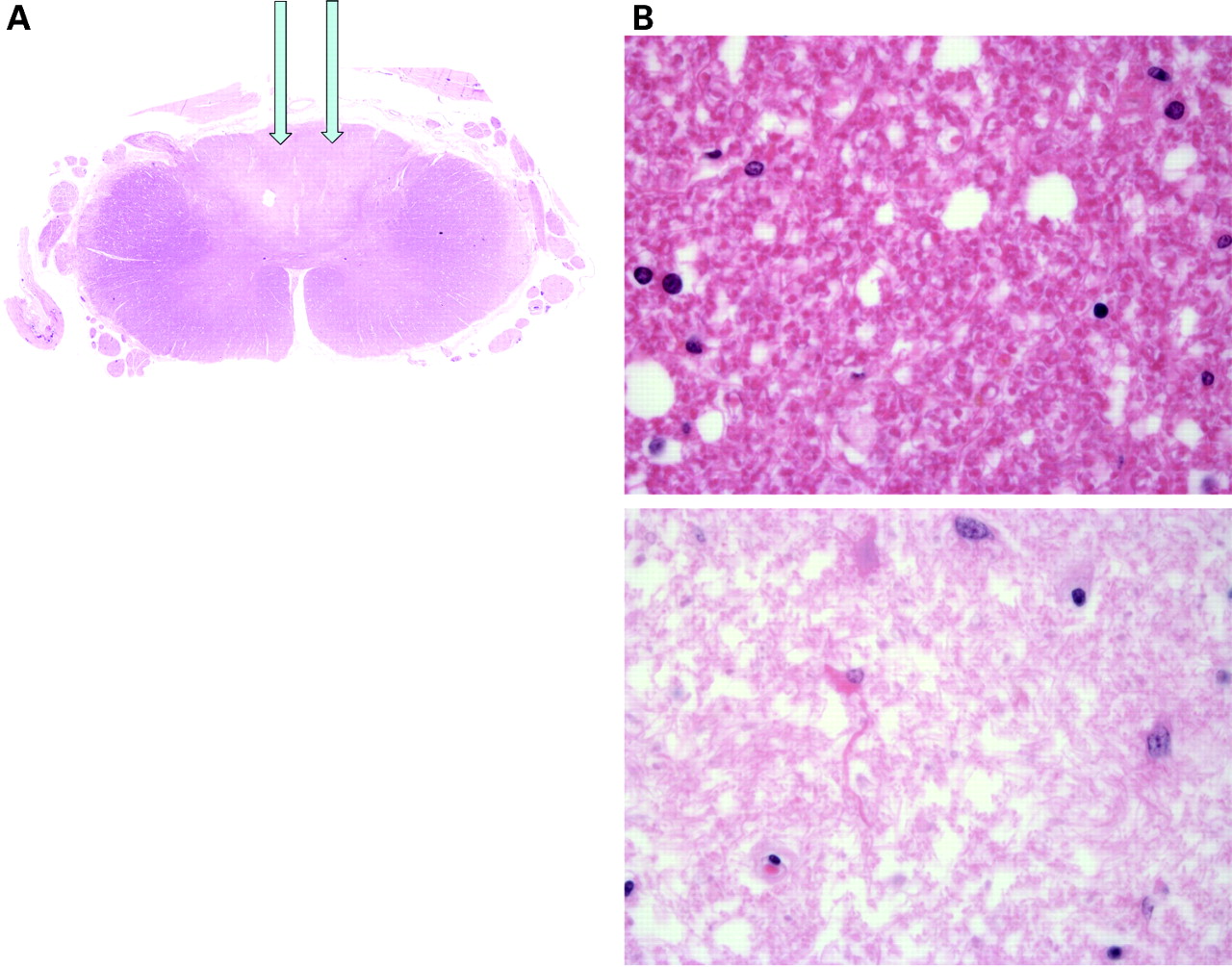
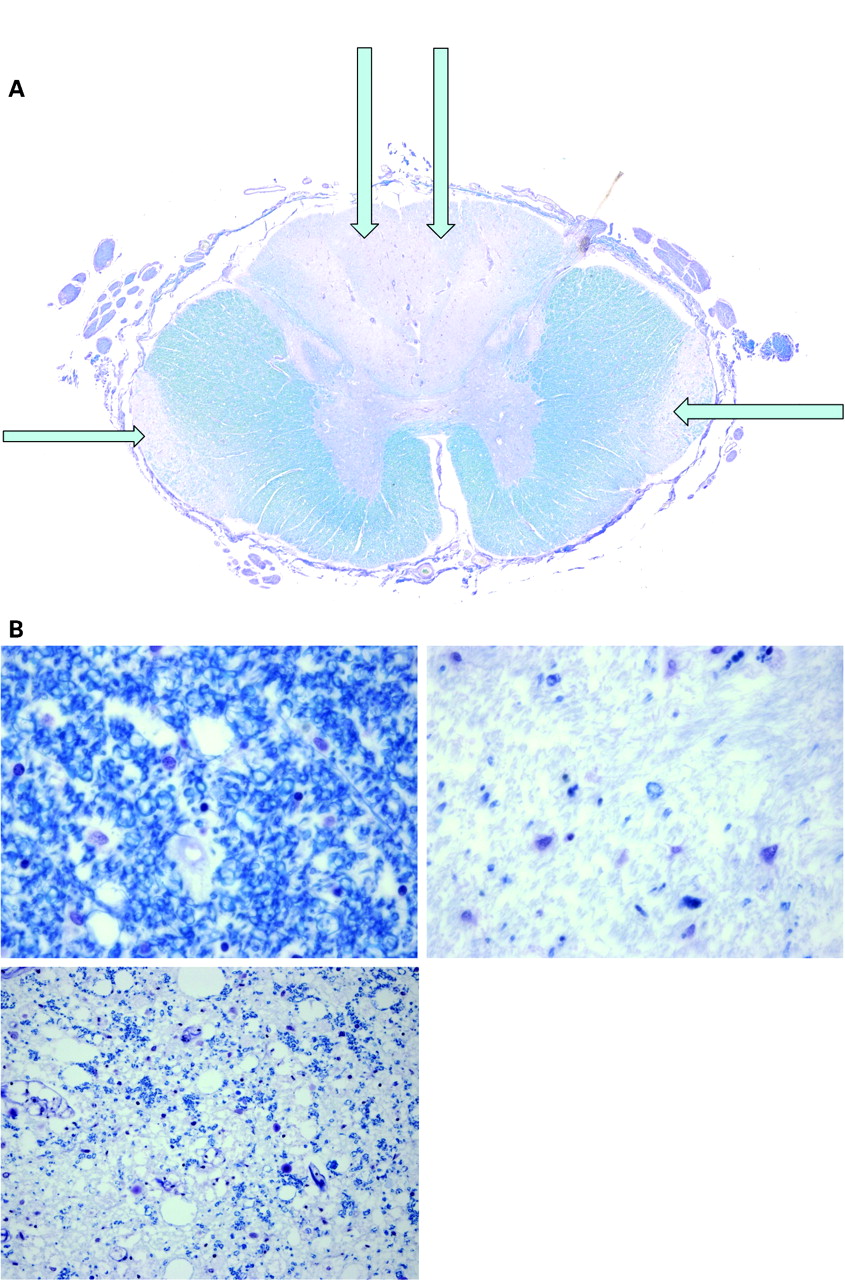
At post mortem the cause of death was bronchopneumonia. The other significant pathology included significant jejunal diverticulosis and pathology in the central nervous system, predominantly involving the spinal cord which was examined at multiple levels. Haematoxylin–eosin sections of the cord revealed pallor of the dorsal columns which was highlighted with a myelin stain. The appearances were of demyelination consistent with subacute combined degeneration of the cord (fig 4). At high power, Luxol Fast Blue stained sections demonstrated pallor of the dorsal columns and lateral funiculi with a reduction in the number of myelin sheaths (fig 5). This was an active disease process. Immunohistochemistry with the macrophage marker CD68 highlighted phagocytic activity indicating ongoing myelin sheath degeneration.
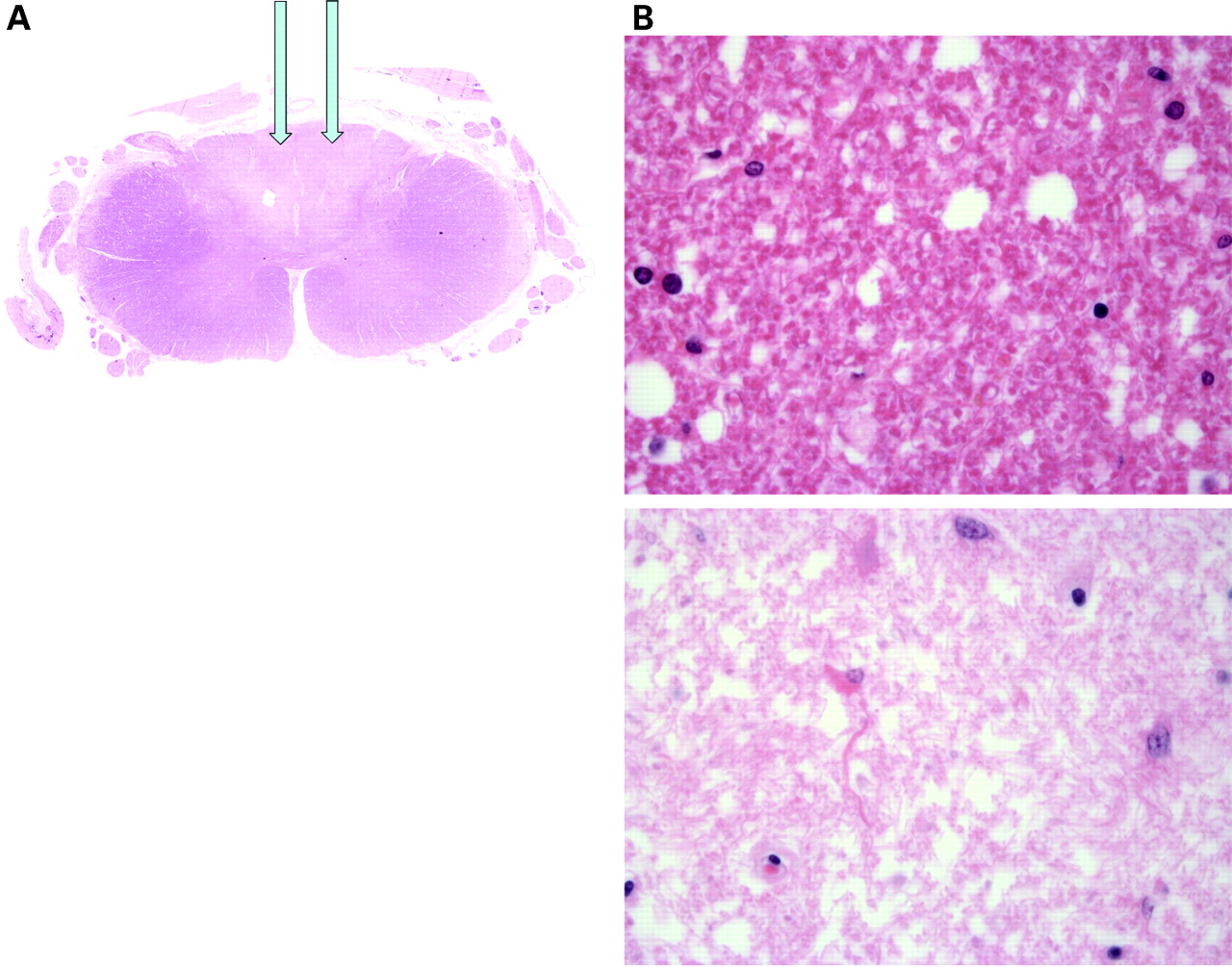
(A) Haematoxylin–eosin (H&E) section of the spinal cord showing abnormal regions which when stained for myelin demonstrated demyelination consistent with subacute combined degeneration of the cord with pallor of the dorsal columns (arrows). (B) H&E stained section (×60) showing relatively normal cord (above) and abnormal area (below).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) At high power, Luxol Fast Blue stained sections demonstrate pallor of the dorsal columns (arrows) and lateral funiculi (arrows) with a reduction in the number of myelin sheaths. (B) Luxol Fast Blue stained sections from the cord (×60), showing varying degrees of damage.
Therefore, initial assessment suggested the diagnosis was straightforward. The patient had jejunal diverticulosis with stasis and bacterial overgrowth, resulting in vitamin B12 deficiency, degeneration of the dorsal columns and lateral funiculi, resulting in subacute combined degeneration of the cord secondary to the vitamin B12 deficiency. However, vitamin B12 deficiency was diagnosed and treated from 1995 and, despite repeatedly normal B12 levels, he continued to deteriorate neurologically in the 18 months prior to death. Although the pathological features are typical of subacute combined degeneration of the cord, there are a number of other possible causes of this clinical syndrome, such as AIDS associated vacuolar myelopathy, transverse myelitis associated with lupus erythematosus, demyelination such as in Devic’s disease and non-B12 deficient subacute combined degeneration due to hypocupraemia. With the exception of hypocupraemia the histopathological appearances do not support any of these differential diagnoses.
The syndrome of copper deficiency myelopathy mimics vitamin B12 deficiency myelopathy, both clinically and radiologically. The reported associations include gastric surgery, hyperzincaemia, malabsorption syndromes and parenteral nutritional deficiency in copper.
Most copper absorption appears to occur in the duodenum, possibly via the transporter protein Ctr1, or other less specific transporters such as Dmt1 (also used by iron). Copper is transported to the blood using ATP7A (Cu-ATPase) which is deficient in Menke’s disease. Most copper homeostasis is undertaken in the liver using ATP7B. Copper is a key constituent of various metalloproteinases such as cytochrome oxidase C and superoxide dismutase and has an important role in mitochondrial metabolism.8
To summarise this case, this patient had a malabsorption syndrome due to jejunal diverticulosis and chronic duodenitis. His myelopathy is unlikely to have been due to proven vitamin B12 deficiency because it was adequately supplemented. Although the biochemistry is not available to confirm this, he is most likely to have had hyocupraemia to account for his neurological syndrome and physical decline which resulted in his immobility and ultimate pneumonia.
DISCUSSION
Professor Charles Warlow: We can’t be absolutely sure there was really nothing else going on in this case. The patient presented at a time when the neurological syndrome was not well known among British neurologists. Even if the diagnosis had been made, the chances of him making a recovery were very small.
Member of the audience: Abnormalities were noted on neuroimaging within the brainstem. Was this examined pathologically?
Dr Colin Smith: No, but similar findings to those found in the spinal cord are well described in the literature in copper deficiency.
Dr Ravi Malhotra: Were the dorsal root ganglia examined?
Dr Colin Smith: A number were sampled and did not show any abnormalities.
Member of the audience: Are copper levels in the liver useful in this situation?
Dr Colin Smith: In this patient, the postulated deficiency was due to malabsorption from the gastrointestinal tract, whereas in Wilson’s disease there is a defect with transport out of the liver, so I don’t think they would be relevant here.
Dr Kevin Talbot’s clinical diagnosis
Jejunal diverticulae causing malabsorption of copper to produce subacute combined degeneration of the spinal cord.
Dr Colin Smith’s pathological diagnosis
Jejunal diverticulosis and chronic duodenitis, with myelopathy due—probably—to malabsorption of copper.
Linked Articles
- Editor’s choice
Other content recommended for you
- Nitrous oxide-induced subacute combined degeneration of the cord: diagnosis and treatment
- Helicobacter pylori associated vitamin B12 deficiency, pernicious anaemia and subacute combined degeneration of the spinal cord
- Functional vitamin B12 deficiency
- A man with tingling fingers
- Neurology and the gastrointestinal system
- Clinical assessment of the sensory ataxias; diagnostic algorithm with illustrative cases
- The neurology of enteric disease
- Common variable immunodeficiency leading to spinal subacute combined degeneration monitored by MRI
- Early treatment causes clinicoradiological reversal of myelopathy due to vitamin B12 deficiency
- Progressive myeloneuropathy with symptomatic anaemia