Article Text

Statistics from Altmetric.com
Introduction
The distinctive patterns of myelopathy (disorder of the spinal cord) and radiculopathy (disorder of spinal roots) are a direct consequence of the striking anatomy of the spinal cord:
▶ its near cylindrical, segmental structure of great length (42–45 cm in adults)
▶ the marked proximity of ascending and descending long tracts within the confines of a narrow cross sectional area (the maximum circumference of the cervical enlargement of the cord is approximately 38 mm)
▶ enclosure by meninges and vertebral column
▶ vulnerable blood supply.
Having established that a patient's clinical presentation localises to the spinal cord and/or roots, clues to the pathological diagnosis emerge from the timing of the symptoms (table 1), as is usually the case in neurology.
Speed of onset and likely cause of myeloradiculopathy
Neuroanatomy and specific syndromes
Spinal cord
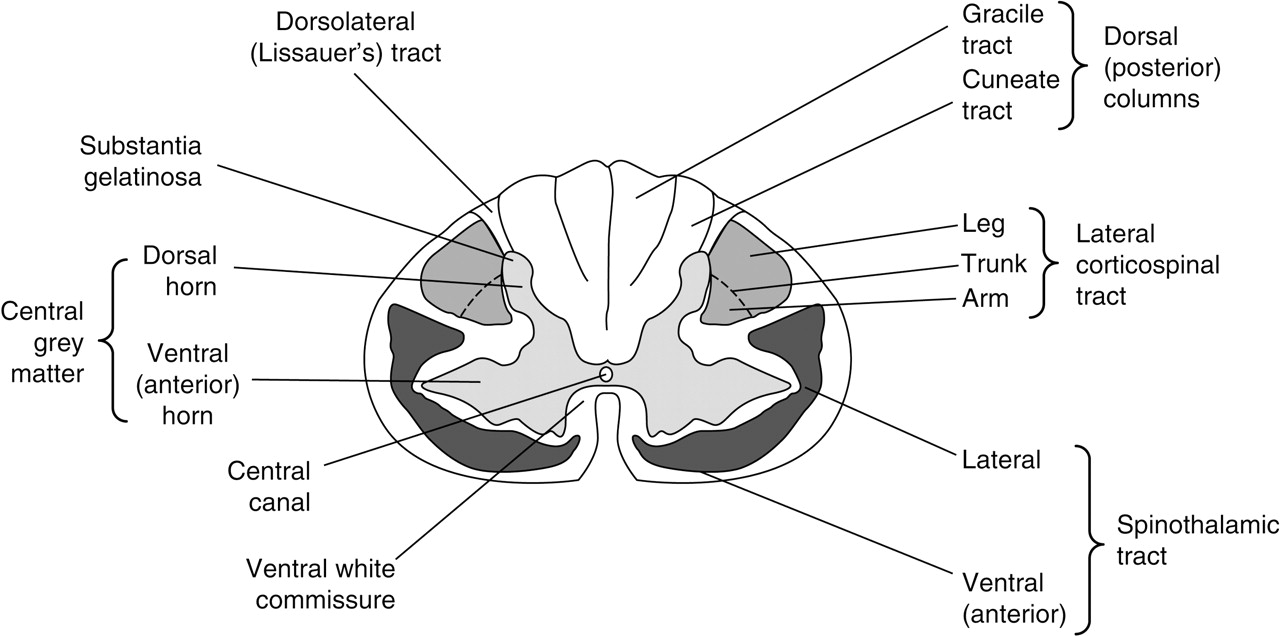
The relationships of the white matter tracts to one another and to the central grey matter are most easily appreciated in transverse section (figure 1). Within the major pathways, there is a relatively orderly somatotopic arrangement:
▶ In the spinothalamic tracts, the most superficial fibres are related to the sacral dermatomes. These overlie the lumbar fibres, with thoracic still deeper in the tracts, and the cervical fibres closest to the central grey matter. Pain and temperature sensation are conveyed in the lateral spinothalamic tracts whereas touch and pressure pathways are anterior (ventral).
▶ In the posterior (dorsal) columns, fibres from the lower limbs ascend medially in the gracile tracts, those from the upper limbs are lateral in the bulkier cuneate tracts. Pathways for position and touch sensation generally lie deep to those for vibration and pressure.
▶ In the lateral corticospinal tracts, descending fibres to the upper limbs are medial to those to the lower limbs.

Transverse section of the spinal cord in the mid-cervical region. Many ascending and descending tracts have been omitted to emphasise the most clinically relevant.
The organisation is similar in the central grey matter of the cord where the neuronal cell bodies are arranged in 10 layers or ‘nuclei’ (Rexed's laminae):
▶ The laminae in the dorsal horns are where the primary cutaneous afferent fibres terminate having reached the cord via the dorsal roots of the spinal nerves. They then synapse with second order neurons in the substantia gelatinosa of the dorsal horns, directly or through interneurons. These second order neurons decussate in the ventral white commissure and ascend in the contralateral spinothalamic tract (this pathway may be contrasted with that for proprioceptive and exteroceptive information ascending in the dorsal columns where the central axons of the primary afferents remain ipsilateral and may not synapse until reaching the gracile and cuneate nuclei in the medulla).
▶ The lateral horns of the central grey matter are small projections in the thoracolumbar cord (T1 to L2) which contain the cell bodies of preganglionic sympathetic neurons.
▶ The ventral (anterior) horns are much larger and contain the cell bodies of lower motor neurons. Anterior horn cells are arranged so those whose axons are destined to innervate flexor muscles lie dorsal to those supplying the extensors; lower motor neurons to the limb muscles originate lateral to those destined for the truncal musculature in the ventral horns.
These microscopic arrangements and relationships all have clinical relevance when specific cord syndromes are considered:
Extrinsic compression
A lesion gradually compressing the spinal cord can produce weakness below the level of the lesion (spastic paraparesis, spastic tetraparesis, or upper motor neuron signs in the legs with a combination of upper and lower motor neuron signs in the arms), sphincter disturbance and sensory loss, ultimately to all modalities. There may be local or radicular pain. The area of cutaneous sensory impairment typically includes the sacral dermatomes because the fibres from this area lie closest to the surface of the cord in the spinothalamic tracts and are therefore most vulnerable to extrinsic compression.
This pattern is the opposite of that in intrinsic cord lesions, where there may be sacral sparing.
The upper limit of the impaired sensation—the sensory level—may correspond to the level of the compressive lesion, but not always. Thus a sensory level at T10, for example, implies that the lesion is at T10 or above. The explanation is again related to the lamination of the spinothalamic tracts. Early in its evolution, a compressive lesion at the cervicothoracic junction (C8/T1), for example, may only cause relatively mild cord impingement, damaging the outermost layers of the spinothalamic tracts—that is, those relating to the sacral and lumbar dermatomes. In time, however, as the compression worsens and deeper fibres within the tracts become affected, the sensory level ascends towards where the lesion actually is—at T1. The upper level of the sensory disturbance therefore depends on the stage of evolution of the lesion. Indeed patients may describe this process in their history, saying that the numbness began in their feet, gradually ascended to the knees and eventually to their trunk. This evolving pattern is of great practical importance in the investigation and management of acute cord lesions. If a patient with a sensory level at T10 has only the lower thoracic cord imaged, a surgically treatable lesion further up the spine may be missed.
Acute complete transverse lesions
Acute complete transverse lesionsproduce an extreme version of the pattern seen with extrinsic cord compression of more gradual onset; complete paralysis and sensory loss to all modalities below the level of the lesion, and loss of sphincter control. Immediately after the injury, however, there is a period of spinal shock typically lasting 1–2 weeks or sometimes longer when the motor signs are paradoxical, with flaccid paralysis and absent tendon reflexes. This is why acute cord lesions enter the differential diagnosis of the Guillain–Barré syndrome. Upper motor neuron signs only develop after spinal shock has resolved. A similar sequence of events is seen with sphincter function after severe spinal cord injury, with an initially atonic bladder and bowel, later becoming spastic. Acute complete transverse lesions can be traumatic or non-traumatic (eg, vascular).
Central cord syndrome
Some patients with limited trauma to the cervical cord may have disproportionate damage to the central grey matter, with relative sparing of the long tracts; the dominant clinical findings are then of lower motor neuron signs in the arms, with variable sensory loss. Mild, reversible trauma, often in the context of pre-existing cervical spinal stenosis, may result in numb, clumsy or ‘burning’ hands without weakness or long tract features, the pathology presumably residing in or near the dorsal horns.
A similar pattern, albeit much more chronic, is seen in syringomyelia where a cavity develops within the cord, usually starting close to the cervicothoracic junction and gradually extending rostrally and caudally, in association with a Chiari malformation. The cavity affects the decussating spinothalamic fibres in the ventral white commissure, producing a characteristic suspended or cape-like loss of pain and temperature sensation, with upper and lower sensory levels. Eventually, the long tracts are involved, so the patient has a combination of lower motor neuron signs in the upper limbs and upper motor neuron signs in the lower limbs, but a degree of dissociated anaesthesia persists—ie, with sparing of dorsal column function.
A syrinx may also occur in association with intramedullary spinal tumours and after trauma and other causes of extrinsic cord compression.
Ventral cord syndrome
The blood supply of the spinal cord is vulnerable to hypotension. The anterior spinal artery supplies the anterior two-thirds of the cord and is a functional end artery. The cord's longitudinal arteries (one anterior and two posterior) are insufficient to provide its entire transverse or longitudinal needs and are supplemented by segmental medullary feeder vessels. Of these, the largest is the artery of Adamkiewicz which arises from a segmental vessel of the aorta, usually between T9 and L2 on the left, and this can be responsible for most of the supply to the lower cord. The upper to mid thoracic cord is distant from this and other feeders, hence it is at particular risk of ischaemia at times of hypotension. Anterior spinal artery infarction produces paraplegia or tetraplegia, loss of pain and temperature sensation below the lesion, with relative sparing of dorsal column function (because the posterior one-third of the cord is supplied by the posterior spinal arteries).
Dorsal cord syndrome
While the dorsal columns can sometimes be selectively damaged by vascular occlusion, the more usual pathology (in combination with that of the lateral corticospinal tracts) is metabolic or inflammatory (see below).
Hemicord syndrome
The typical features of the Brown–Séquard syndrome, when the lesion is confined to one side of the cord, arise because of the decussation of spinothalamic fibres in the ventral white commissure. As a result, corticospinal tract and dorsal column function are impaired on the side of the lesion whereas pain and temperature sensation are affected on the opposite side of the body. Patients may also have a narrow band of spinothalamic sensory loss, sometimes accompanied by pain, on the side of the lesion and close to its level, caused by damage to fibres which have not yet decussated to join the contralateral spinothalamic tract. Originally described (and still seen) in the context of trauma, any asymmetrical cord lesion, such as a plaque of demyelination, can cause this, albeit often partial.
Further reading
Anatomical source texts
▶
▶
Commoner causes and complications of myelopathy; some of the diseases mentioned in this article are described in more detail in other contributions to the ‘Bare essentials’ series
Rarer causes
▶ OpenUrlAbstract/FREE Full Text
Management and rehabilitation
▶
Conus lesion
A lesion at the lower end of the spinal cord can produce a mixture of upper and lower motor neuron signs in the legs, typically absent ankle reflexes (due to damage to the reflex arc) with upgoing plantar responses (due to damage to the corticospinal tract). Weakness in the lower limbs is mild or absent but there is profound sphincter disturbance and saddle anaesthesia.
Foramen magnum lesion
Lesions of the high cervical cord can produce confusing symptoms and signs. Weakness may begin in one arm and then spread in a clockwise or anti-clockwise fashion to the ipsilateral leg, then the contralateral leg and finally to the contralateral arm. Lower cranial nerve palsies may be present, along with downbeating nystagmus. Pain and paraesthesiae may affect the upper limb first showing motor involvement. For poorly understood reasons, possibly vascular in basis, high cervical cord lesions sometimes produce physical signs in the arms which suggest lower motor neuron involvement, with distal wasting and weakness, and depressed tendon reflexes.
Spinal roots
The ventral and dorsal roots at each spinal level unite to form mixed (sensory and motor) spinal nerves. The point of union is in or close to the intervertebral foramen. Spinal nerves and roots are numbered according to the adjacent vertebrae (figure 2). Thus in the cervical region, the nerve is the same number as the vertebra below it (eg, the right and left C7 nerves emerge from the spine at C6/7). The C8 nerves lie between the C7 and T1 vertebrae so spinal nerves below this level exit below the vertebrae with which they share a number. However, a disc prolapse in the lumbar region will still generally impinge on the root(s) with the same number as the vertebra below (because of the three-dimensional arrangement of the roots in the lower spinal canal). A lateral disc prolapse at L4/5, for example, will generally cause an L5 root lesion. It will only involve the L4 nerve if there is a far lateral protrusion.

Diagram showing the relationship of the spinal cord segments and spinal nerves to the vertebrae. (Reproduced with permission from Ginsberg L. Lecture notes on neurology, 9th Edn. Wiley-Blackwell, 2010.)
A spinal cord segment is defined by the zone of attachment of the rootlets of a pair of spinal nerves (right and left, dorsal and ventral roots, 6–8 rootlets per root). As the cord occupies only the upper two-thirds of the spinal canal, there will necessarily be non-correspondence between the cord segments and the vertebrae (figure 2). This divergence becomes more marked the more caudal the segment, so that the lower roots have a longer intraspinal course than those arising more rostrally. Ultimately, with the spinal cord ending around L1/2, the neural elements in the lowest part of the theca consist solely of the roots forming the cauda equina, crowded around the filum terminale.
Monoradiculopathies
Monoradiculopathies are usually compressive but can be inflammatory or infective. Their clinical features comprise radicular pain, dermatomal sensory loss, weakness and sometimes loss of reflex(s). Radicular pain may be perceived in the relevant myotome, rather than the dermatome—a useful practical point. For example, C5 pain may localise medial to the scapula, in the rhomboids. Testing the strength of particular muscles may indicate which root is affected—segment-pointer muscles (table 2).
Segment-pointer muscles*
Polyradiculopathies
Polyradiculopathies may be compressive, inflammatory or infiltrative. A compressive lesion of the cauda equina (caused by central intervertebral disc prolapse or tumour) may be indistinguishable from a conus lesion but there will be no upper motor neuron signs. Cauda equina syndrome is also typically more painful than a conus lesion, and more likely to cause lower limb weakness.
Causes of myelopathy (and radiculopathy)
Spinal cord and root disease may first be divided into extrinsic (usually compressive) and intrinsic causes, then the familiar ‘surgical sieve’ can be applied (table 3).
Causes of myelopathy
Genetic
▶ Hereditary spastic paraplegia is a neurodegenerative process primarily affecting upper motor neurons, rather than a disease of the spinal cord per se. It can resemble a ‘true’ myelopathy clinically, however, even in its relatively uncomplicated forms where there may be sphincter and sensory involvement. The pattern of inheritance is most commonly autosomal dominant but can be autosomal recessive or sex linked.
▶ Adrenomyeloneuropathy is one presentation of X linked adrenoleukodystrophy. Males (and occasionally female manifesting carriers) develop a slowly progressive spastic paraparesis in young adulthood, followed by sensory and sphincter involvement. Adrenal failure need not occur. Raised very long chain fatty acid levels are diagnostic in males but mutation analysis may be required in females.
▶ Other leukodystrophies, for example, metachromatic leukodystrophy, can have prominent upper motor neuron features, but the presence of cognitive deterioration and brain MRI changes usually help clarify the diagnosis.
▶ Some of the hereditary ataxias may occasionally present with a predominant spastic paraparesis. In particular, variant patients with Friedreich's ataxia may have lower limb spasticity, pyramidal weakness and retained reflexes. Similarly, some patients with spinocerebellar ataxia type 3 (Machado–Joseph disease) have clinical features resembling hereditary spastic paraplegia early in the course of their illness.
▶ Genetic disorders can also cause extrinsic compressive cord lesions, for example, in the mucopolysaccharidoses where the metabolic defect may lead to spinal dysostosis, dural thickening or atlanto-axial instability.
Congenital (ie, starting during fetal development)
▶ Syringomyelia is probably congenital, in association with a Chiari malformation.
▶ Spinal dysraphism varies in severity from the chance radiographic finding of spina bifida occulta to a full blown lumbar myelomeningocoele with consequent lower cord/cauda equina syndrome and neonatal hydrocephalus. Adults presenting with a partial conus or cauda equina syndrome in association with developmental lumbosacral skin changes (dimples, pits, sinuses, lipomas, haemangiomas or tufts of hair) may have a variety of underlying spinal defects, for example, tethering of the cord, or a lumbar syrinx or lipoma. Further up the spine, a bony spur can separate the cord into two halves—diastematomyelia.
▶ Arachnoid cysts are also probably congenital. Although usually asymptomatic, they can occasionally cause chronic cord compression, sometimes with secondary syrinx formation.
Traumatic
Missile and stab injuries to the spine are encountered in civilian life but the more common trauma is crushing of the cord consequent on fracture–dislocation of the spine, potentially exacerbated by hyperflexion/hyperextension and by any pre-existing narrowing of the spinal canal. Such injuries usually transect the cord with instantly fatal results if the lesion is in the high cervical region, unless there is immediate respiratory support (the respiratory muscles are innervated by motor fibres arising from C3 to the lower thoracic cord). More caudally, the degree of paralysis depends on the level of the lesion but there will generally be a phase of spinal shock followed by the development of hyperreflexia.
Infective
Extrinsic infective processes compressing the cord or cauda equina include epidural abscess, usually due to Staphylococcus aureus (sometimes streptococci and anaerobes). The clinical presentation is with the triad of pain (local, with tenderness and/or radicular), fever and signs of myeloradiculopathy.
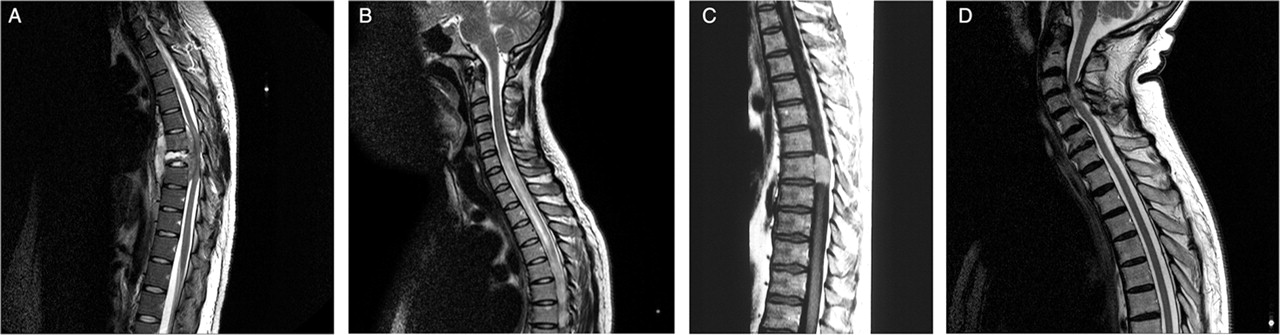
Tuberculous spinal osteomyelitis (figure 3A) is encountered particularly in at risk groups (those in or from developing countries, or immunocompromised individuals) and leads to neural compression which may be associated with spinal deformity, ultimately the angular gibbus of Pott's disease of the spine.

{kind=link}
{kind=link}
{kind=link}
Spinal MRI. (A) Tuberculous spinal osteomyelitis and discitis—sagittal T2 weighted image of the thoracic spine showing destruction of the T8 vertebral body and extradural compression of the spinal cord. (B) Neuromyelitis optica—sagittal T2 weighted image of the cervicothoracic spine showing longitudinally extensive transverse myelitis, involving much of the cord caudally from C5. (C) Spinal meningioma—sagittal T1 weighted image of the thoracic spine post-gadolinium. The lesion was isointense with the spinal cord on the pre-contrast images but can be seen to enhance avidly post-gadolinium. (D) Spondylotic compression at C3/4—sagittal T2 weighted image of the cervicothoracic spine. There is anterior and posterior cord impingement, resulting in damage to its parenchyma (myelomalacia), shown as high signal within cord substance at the point of maximal compression. (Figure 3C is reproduced with permission from Ginsberg L. Lecture notes on neurology, 9th Edn. Wiley-Blackwell, 2010.)
Intrinsic infective myelopathies can be acute or chronic:
▶ Acute infective myelitis is less common in developed countries than a post-infective process (see below). It may be viral (zoster, herpes simplex, HIV at seroconversion), bacterial (tuberculosis, secondary syphilis) or, rarely, parasitic (schistosomiasis).
▶ Chronic infective myelopathies include the vacuolar myelopathy of advanced HIV infection. Another chronic retroviral myelopathy is associated with human T cell lymphotropic virus infection (tropical spastic paraparesis, human T lymphotropic virus type 1 associated myelopathy) which is endemic in the Caribbean, sub-Saharan Africa and the Far East. Transmission is vertical, sexual or through blood transfusion. Only a small minority of infected individuals develop neurological problems following an incubation period of many years. The clinical picture is of a progressive spastic paraparesis (usually slow) with prominent sphincter involvement and painful paraesthesiae in the legs.
Infective radiculopathies include herpes zoster and also the meningoradiculopathy of Lyme disease. A polyradiculitis of the cauda equina caused by cytomegalovirus occurs in AIDS patients with very low CD4 cell counts.
Inflammatory and demyelinating
Multiple sclerosis (MS) is the most common cause of a spastic paraparesis in young adults in temperate zones. Although a complete transverse myelitis may occur, the clinical features of a cord relapse of MS are typically asymmetrical, reflecting the likely irregular disposition of the causative plaques. The Lhermitte phenomenon is helpful in the diagnosis of MS, but not pathognomonic, because it also occurs in compressive cervical cord lesions and vitamin B12 deficiency. The Uhthoff phenomenon is also useful, but may be more subtle than the patient simply complaining of worsening symptoms after a hot bath. Thus patients with spinal cord demyelination may describe exercise induced deterioration in their walking, mimicking vascular or compressive spinal lesions.
Theoretically, there is no limit to the length of a spinal plaque of MS. However, very long intrinsic cord lesions identified on MRI are increasingly being associated with other causes. This has led to the emergence of the awkwardly named but clinically useful concept of longitudinally extensive transverse myelitis (extending at least three vertebral segments, figure 3B). This can be seen in neuromyelitis optica (NMO, Devic's disease) where the clinical cord syndrome is usually much more severe than in MS.
Post-infective or post-immunisation transverse myelitis may be a forme fruste of acute disseminated encephalomyelitis. The typical presentation is with a monophasic acute or subacute transverse myelopathy a few days or weeks after an infection or immunisation. The initial trigger may be relatively trivial, for example, an upper respiratory tract infection. Often, no specific organism is identified but recognised associations include Epstein–Barr virus, varicella zoster virus and Mycoplasma. Spinal MRI may show a longitudinally extensive transverse myelitis and the CSF is usually moderately lymphocytic.
Various systemic inflammatory and autoimmune diseases have occasionally been linked to cord syndromes that resemble MS. The diagnosis is relatively straightforward if the patient already has clinical and/or paraclinical features of the systemic disease, as may occur in sarcoidosis and (rarely) Behçet's disease. A diagnosis of sarcoidosis is harder to reach if there are no systemic features. The situation is more controversial for systemic lupus erythematosus and Sjögren's syndrome. Again, there is some diagnostic security if there are systemic features of the autoimmune disease. However, if the only evidence of lupus or Sjögren's is serological, the patient may in fact have NMO with non-pathogenic antibodies.
Systemic inflammatory disorders may also cause extrinsic cord lesions. The most notorious is atlanto-axial subluxation, a rare complication of rheumatoid arthritis, where the combination of a build-up of inflammatory tissue and weakening of ligaments puts the patient at risk of a catastrophic compressive foramen magnum lesion.
Neoplastic
Spinal tumours are classified as extradural, extramedullary–intradural and intramedullary:
▶ Extradural tumours include metastases to the vertebrae (typically breast, lung, prostate, thyroid and kidney cancers) and other bony malignancies (myeloma, lymphoma). These lesions impinge on the cord, roots or cauda equina by invading the epidural space, or by causing vertebral collapse. Patients present with acute cord compression, back pain and tenderness, but the history can often be traced back days or even weeks. Sometimes, malignant disease can spread through the epidural space, ‘picking off’ roots and compressing the cord at multiple levels, without involving the vertebrae. This applies particularly to lymphoma and prostatic carcinoma. The result can be a confusing mixture of upper and lower motor neuron signs, even mimicking motor neuron disease. Non-malignant extradural tumours include chordomas and lipomas.
▶ Extramedullary–intradural tumours lie on the surface of the cord, having arisen from the meninges or a root, mainly meningiomas and neurofibromas. Spinal meningiomas (figure 3C) have a much more marked female preponderance than their intracranial counterparts. Neurofibromas may occur in isolation or in the context of neurofibromatosis. They are more likely to generate radicular symptoms than meningiomas.
▶ Intramedullary tumours lie within the spinal cord, sometimes with a syrinx. They are very rare and include astrocytomas, ependymomas, haemangioblastomas and metastases.
An acute necrotising myelopathy may occur as a paraneoplastic phenomenon, most commonly with small cell lung cancer. The cord syndrome may antedate the discovery of the cancer by many months. Antineuronal antibodies (anti-Hu) are usually present.
Vascular
Like the brain, the cord can be affected by both haemorrhagic and ischaemic vascular events. Bleeding into the epidural or subdural space, for example, through anticoagulant excess, will cause an acute compressive transverse myelopathy. Haemorrhage within the cord (haematomyelia) may be due to an underlying arteriovenous or cavernous malformation.
Ischaemic cord infarction most commonly affects the anterior spinal artery territory, producing a ventral cord syndrome of abrupt and painful onset. Causes include atherosclerosis of feeder vessels (in particular, the artery of Adamkiewicz), aortic dissection, and aortic surgery (especially if complicated by hypotension). Rarer possibilities are thrombosis of the anterior spinal artery itself, vasculitis, infective endocarditis, decompression sickness and fibrocartilaginous embolism.
Spinal dural arteriovenous fistula is uncommon but potentially treatable. Patients are usually male and middle-aged or elderly, presenting with a subacute or chronic, stepwise, fluctuating deterioration in gait, with pain in the back or leg, sensory disturbance and ultimately sphincter involvement. Exercise may trigger deterioration. On examination, a combination of upper and lower motor neuron signs may be seen. Although the fistula is usually in the lower cord, the sensory level may be several segments higher because of ascending venous congestion. Spinal MRI may show only subtle signal change in the lower cord and conus or an enlarged draining vein, although spinal MR angiography may be more informative. Confirmation of the diagnosis ultimately requires selective catheter angiography. The fistula may be amenable to embolisation or surgical occlusion.
Metabolic and toxic
As the name implies, subacute combined degeneration of the cord, secondary to vitamin B12 deficiency, is characterised by a combination of dorsal column and corticospinal tract dysfunction, usually evolving over the course of a few weeks. There is also a significant component of peripheral neuropathy (hence the coexistence of lower motor neuron with the upper motor neuron signs from the myelopathy—absent ankle reflexes and extensor plantar responses) and, later, optic neuropathy and cognitive impairment. Vitamin B12 deficiency is remarkable as one of the few causes of peripheral neuropathy where the hands may be affected before the feet, but this is largely because of the coexistent myelopathy; spinal MRI shows signal change in the dorsal columns of the cervical cord. The neurological complications of vitamin B12 deficiency may occur in the absence of any haematological disorder, even with a normal mean corpuscular volume. Investigation should include measurement of serum methylmalonic acid and homocysteine as well as vitamin B12 levels.
Copper deficiency has been recognised recently as a cause of a subacute combined degeneration picture. At risk patients include those who have undergone partial gastrectomy or gastric bypass surgery (hence impairing copper absorption) and individuals with excessive zinc intake (zinc competing with copper for absorption in the proximal duodenum). The diagnosis is confirmed by very low serum copper and caeruloplasmin levels. The haematological findings of copper deficiency are different from vitamin B12 deficiency, sideroblastic anaemia with neutropenia.
Chronic liver failure may lead to a range of neurological deficits, including a slowly progressive myelopathy.
Toxic causes of myelopathy include:
▶ Nitrous oxide inhalation producing clinical features resembling subacute combined degeneration. Patients having multiple anaesthetics are at risk, as are dentists and veterinary surgeons who self-administer for recreational purposes.
▶ Oil based contrast medium (Myodil), previously used for myelography, causes spinal arachnoiditis with consequent cord and cauda equina damage. Arachnoiditis can also develop after spinal surgery.
▶ Radiation—a slowly progressive myelopathy may occur months or years after radiotherapy.
▶ Dietary toxins—lathyrism from consumption of the chickling vetch Lathyrus sativus, konzo from cassava.
Degenerative
Cervical spondylotic myelopathy is the most common cause of a spastic paraparesis in elderly patients. The extrinsic compression arises from a combination of degenerative disc disease, osteophyte formation and ligamentous hypertrophy, leading to myelomalacia (figure 3D). A single level in the lower or mid cervical region may be affected (as shown in figure 3) but often there is multilevel disease. The clinical features are of a spastic paraparesis, with or without sphincter and sensory involvement, in association—but not always—with neck pain and stiffness, and radicular arm pain. Upper limb signs may be a mixture of upper and lower motor neuron. It is often possible to discern the site of the lesion clinically from the presence of a ‘reflex level’. Thus an absent or inverted biceps reflex, with brisk reflexes below, implies myeloradiculopathy at C5.
Lumbar spondylosis, frequently superimposed on a congenitally stenotic spinal canal, may cause chronic cauda equina compression. Patients present with back and leg pain, with sensory and motor symptoms in the lower limbs, which may be exercise induced—intermittent claudication of the cauda equina. Neurogenic claudication is typically triggered by walking and relieved by sitting or leaning forwards for 5–15 min, considerably longer than is required to relieve claudication from peripheral vascular disease. This relationship of symptoms to posture is thought to reflect the cross sectional area of the lumbar canal, which is least on standing and walking, hence resulting in maximal compression of the cauda equina and its blood supply.
Disc protrusions usually occur spontaneously, in the context of disc degeneration, rather than being linked to a specific traumatic event. Lateral disc protrusions typically cause a cervical or lumbar monoradiculopathy. Central disc prolapse in the lumbar region may cause an acute cauda equina syndrome, a neurosurgical emergency.
Motor neuron disease (amyotrophic lateral sclerosis) occasionally presents as a spastic paraparesis—an intrinsic (neuro)degenerative cause of myelopathy. In the absence of lower motor neuron signs, the diagnosis may be supported by detecting subclinical evidence of denervation on electromyography. Primary lateral sclerosis is a virtually pure symmetrical spastic paraparesis, progressing to tetraparesis then pseudobulbar palsy, with sphincter involvement and emotional lability only occurring late in the course of the disease. Primary lateral sclerosis has a much better prognosis than amyotrophic lateral sclerosis. The diagnosis rests on a minimum disease duration of 3 years and the absence of pointers to other causes (eg, no family history to suggest hereditary spastic paraplegia, no oligoclonal bands in CSF to suggest primary progressive MS).
Principles of management
Investigation
The key investigation for a patient presenting with a myelopathy or lumbar polyradiculopathy is spinal MRI to identify or exclude cord or cauda equina compression. The decision to image a patient with a cervical or lumbar monoradiculopathy depends on the severity and duration of symptoms. If cord or cauda equina compression has been excluded, further investigation will be determined by the patient's clinical presentation and spinal MRI findings (table 4).
Investigation of myelopathy
Surgery
The need for urgent surgery is not controversial for patients with cord or cauda equina compression from:
▶ benign extradural or extramedullary–intradural tumour
▶ epidural abscess
▶ epidural haematoma
▶ acute central disc prolapse.
Indeed, the last three diagnoses are neurosurgical emergencies. Malignant extradural tumours respond as well to radiotherapy with dexamethasone as they do to surgery.
Surgery for severe trauma should be restricted to spinal stabilisation where there is any risk of further injury, and to aid early rehabilitation. There is no justification for prolonged courses of corticosteroids but methylprednisolone within 8 h of injury may have some modest benefit.
Less clearcut is the surgical management of chronic spondylotic compression, where there is a serious lack of high quality evidence:
▶ Patients with cervical spondylotic myelopathy are often very elderly, with significant comorbidities. The course of the neurological impairment is not necessarily progressive. However, with advancing myelopathy and deteriorating disability, in a patient fit for surgery, there is a case for decompression, mainly to prevent further deterioration (any improvement being a bonus). The posterior operation of decompressive laminectomy may be preferred if multiple levels are affected. Anterior surgery (eg, Cloward's procedure) is used for predominantly discogenic disease.
▶ Many patients presenting with radicular pain from a lateral disc prolapse in the cervical or lumbar region are satisfactorily managed without surgery (see below). Operative intervention should be contemplated only after a failed trial of conservative therapy of at least 6 weeks, but sometimes sooner if there are significant symptoms and signs of root compression.
Medical treatment
Some medical causes of myelopathy demand treatment as urgently as those amenable to surgery, to minimise irreversible cord damage, for example, vitamin B12 replacement for subacute combined degeneration or plasma exchange for a relapse of NMO.
Most patients with acute radicular pain from a lateral cervical or lumbar disc prolapse are managed successfully with analgesia, maintenance of mobility and, if necessary, the involvement of a multidisciplinary pain management team. Epidural and root directed injections of corticosteroids are of uncertain benefit but are sometimes used with the aim of delaying or avoiding surgery.
Rehabilitation
Specialist spinal units have revolutionised the prognosis for survival of patients with severe cord injury. Their environment promotes the attention to detail required to prevent potentially life threatening complications, including venous thromboembolism, pressure sores and urinary tract damage. Other important areas of management for paraplegic and tetraplegic patients are:
▶ prevention of contractures
▶ control of spasticity
▶ bladder, bowel and sexual function
▶ maintenance of nutrition
▶ psychological stress.
Educating and hence empowering patients in the management of their own condition is vital.
Autonomic dysreflexia is a particular complication of cervical and high thoracic spinal cord injuries. It is a reflex response to a noxious stimulus, such as bladder distension, urinary tract infection or lower body trauma. The response manifests as sympathetic overactivity with severe headache and hypertension, the latter occasionally fatal. Prevention may involve the judicious use of α-adrenergic blocking antihypertensive drugs, as well as avoidance of potential precipitants. Acute treatment of autonomic dysreflexia includes sitting the patient upright, removing the precipitant and control of hypertension, for example, with sublingual nifedipine.
Conclusions
▶ The most important investigation for a patient presenting with a myelopathy or lumbar polyradiculopathy is spinal MRI, to detect or eliminate spinal cord or cauda equina compression.
▶ The most common cause of a spastic paraparesis in young adults in temperate zones is MS, whereas cervical spondylotic myelopathy predominates in the elderly.
▶ A spinal epidural abscess presents with the triad of pain (local, with tenderness and/or radicular), fever and signs of myeloradiculopathy.
▶ In myelopathy, a sensory level implies a lesion at or above that level; a motor or reflex level generally has greater localising value.
▶ Spinal shock may last 1–2 weeks after the onset of an acute severe transverse myelopathy and mimic a lower motor neuron syndrome.
▶ The blood supply to the spinal cord is vulnerable to hypotension; the artery of Adamkiewicz, may be responsible for most of the supply to the lower cord.
▶ A Brown–Séquard syndrome, albeit often partial, may be caused by any asymmetrical cord lesion, such as a plaque of demyelination.
▶ The Lhermitte phenomenon occurs commonly in MS, but also with other cervical cord lesions, such as spondylotic compression and subacute combined degeneration.
▶ Longitudinally extensive transverse myelitis, defined as extending for at least 3 vertebral segments, is typical NMO, but also of post-infective transverse myelitis.
▶ Subacute combined degeneration is typically due to vitamin B12 deficiency but a similar clinical picture may be seen with copper deficiency and nitrous oxide inhalation.
▶ Acute cauda equina syndrome from a lumbar central disc prolapse is a neurosurgical emergency; look for ‘red flags’: bilateral leg pain and lower limb motor features, sphincter disturbance and saddle anaesthesia.
▶ Cord or cauda equina compression from a malignant extradural tumour is a neuro-oncological emergency; treatment with radiotherapy and steroids is as good as surgical intervention.
▶ Many patients presenting with radicular pain from a cervical or lumbar lateral disc prolapse are successfully managed conservatively.
▶ Early transfer of patients with severe spinal cord injury to specialist rehabilitation units improves outcome.
Acknowledgments
To Jonathan Rohrer, London who commented on this article, and to Richard Davenport, Edinburgh, who reviewed it.
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.
Other content recommended for you
- Progressive hemiparesis in a 75-year-old man
- Sensitivity of brain MRI and neurological examination for detection of upper motor neurone degeneration in amyotrophic lateral sclerosis
- Myelopathy but normal MRI: where next?
- Myelopathy: chameleons and mimics
- Is it an inflammatory myelopathy?
- Degenerative cervical disc disease causing cord compression in adults under 50
- Mimics and chameleons in motor neurone disease
- Metastatic spinal cord compression
- False localising signs
- SURGICAL DISORDERS OF THE CERVICAL SPINE: PRESENTATION AND MANAGEMENT OF COMMON DISORDERS